
A mild and efficient Zn-catalyzed C-glycosylation: synthesis of C(2)–C(3) unsaturated C-linked glycopyranosides†
Thurpu Raghavender Reddy,
Dodla Sivanageswara Rao and
Sudhir Kashyap*
D-207, Discovery Laboratory, Organic and Biomolecular Chemistry Division, CSIR-Indian Institute of Chemical Technology, Hyderabad-500 007, India. E-mail: skashyap@iict.res.in; Fax: +91-402-716-0387; Tel: +91-402-716-1649
First published on 13th March 2015
Abstract
A highly efficient C-glycosylation method was developed by using catalytic Zn(OTf)2 for the synthesis of 2,3-unsaturated C-glycosides. The scope of the protocol was well illustrated with different substituted glycals and nucleophiles comprising allyltrimethylsilane, triethylsilane, trimethylsilyl cyanide, trimethylsilyl azide, trimethylaluminum, trimethylsilyl phenylacetylene and heterocycles such as thiophene and furan. The present method is mild enough to incorporate C-glycoside linkages in a disaccharide donor as well.
The synthesis of C-linked saccharides, sugar analogs in which glycosidic oxygen is substituted by a carbon atom, is of particular interest due to their usefulness as key intermediates for assembling biologically active molecules1 and natural products like palytoxin, spongistatin, halichondrin, etc.2 Another incentive for focusing on C-glycosides is their stability to hydrolytic cleavage, hence they are employed as potent inhibitors to probe the distinctive biological activities especially studying inhibition and the mechanism of action of carbohydrate-processing enzymes.3 In addition, constructing stereoselective C-glycosidic linkages has unique significance in several modified glycoconjugates such as C-linked α(2,3)sialylgalactose lactone,4 naturally occurring C-nucleosides antibiotics5 and various functionalized β-C-saccharides.6
The synthesis of ‘C-pseudo-glycals’ or 2,3-unsaturated C-glycosides has received wide attention as the C(2)–C(3) double bond present in pyran ring can be further utilized to access valuable sugar derivatives by employing diverse chemical reactions.7 Although, several reagent system have been evolved for the C–C bond formation at the anomeric position of 2,3-unsaturated glycosides, involving (1) Ferrier glycosylation8 (2) Tebbe methylenation and thermal Claisen rearrangement9a (3) Pd-mediated glycosylation9b,c (4) Cu(OTf)2/ascorbic acid catalyzed C-glycosylation of unactivated alkynes9d (5) C-alkynylation with silylacetylene,9e alkynyltrifluoroborates9f and iodoalkynes9g (6) Heck cross-coupling10 of aryl halides,10b,c aryl boronic acids,10d,e benzoic acids,10f and aryl hydrazines10g for the synthesis of aryl-C-glycosides. Despite the stereoselectivity, some of these methods have limitations in terms of substrate scope and restricted to C-alkynylated or C-arylated sugars. Moreover, many of these methods have encountered several disadvantages such as use of strong acidic, toxic, moisture sensitive and oxidizing reagents, use of additive such as strong bases and phosphine ligands, requires high temperature and excess loading of reagents, involves unconventional reaction operation and tedious work-up, provides low anomeric selectivity and poor yields. Besides, the generality and compatibility of reagent system in the presence of other sensitive functionalities remains challenging for carbohydrate chemists.
Owing to the importance of C-linked sugar derivatives, development of an efficient and versatile protocol involving inexpensive, moisture tolerant and eco-friendly chemicals is highly desirable. Indeed, investigating such a reagent system which highlights the general requirement for the synthesis of active pharmaceutical ingredient (APIs) wherein metal-scavenging and residual metal limits under acceptable value is of crucial importance.11 In this context, we have recently demonstrated the utility of non-toxic and mild reagent systems in chemical glycosylation for the synthesis of various functionalized O,N-glycosyl and mannosyl derivatives.12 In continuation of our research in glyco-chemistry, we envisioned the use of Zn(OTf)2 as a valuable reagent for the synthesis of C-glycosides and their derivatives en route to carbohydrate scaffolds containing C-linkage at anomeric position. In this paper, we wish to report a mild and convenient protocol for incorporating C-glycosidic linkages in 2,3-unsaturated-glycosides exploiting the efficiency of Zn(II) triflate catalyst. The present protocol is highly compatible with a wide range of substrates, the stereochemical outcome and activation of different substituted glycal donors highlights the generality and significances of the C-glycosylation.
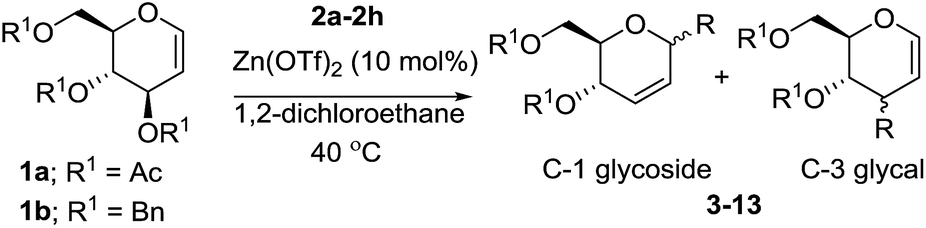
In initial experiments, treatment of preformed dichloromethane solution of 3,4,6-tri-O-acetyl glucal (1a) and allytrimethylsilane (2a) with 10 mol% of Zn(OTf)2 at room temperature provided the desired 4,6-di-O-acetyl-2,3-unsaturated C-allyl glucoside (3) in 54% yields after 16 h. However, the rate of reaction was increased with incremental rise in temperature and 85% conversion of 1a to 3 was realized at 40 °C after 6 h. Surprisingly, switching the solvent system to 1,2-dichloroethane facilitate the reaction and complete conversion was observed in 1 h to obtain 3 in 96% yield with α-anomer as major product (Table 1, entry 1). Further optimization by varying catalytic quantity, temperature or solvent system such as toluene, acetonitrile, THF or diethylether did not lead to any improvement. The structure and stereochemistry of compound 3 was established through spectroscopic analysis and correlated with literature data as well.13a,b
|
|||
|---|---|---|---|
| Entry | Acceptor | Time | Glycosidesb (yield% α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β ratioc) :β ratioc) |
| a All reactions were performed with glucal, 1.2 equiv. acceptor in 1,2-dichloroethane, 10 mol% of Zn(OTf)2 at 40 °C.b Isolated and unoptimized yields.c The C-3 epimeric and anomeric α:β ratios were examined by 1H NMR. |
|||
| 1 |  |
1 h | 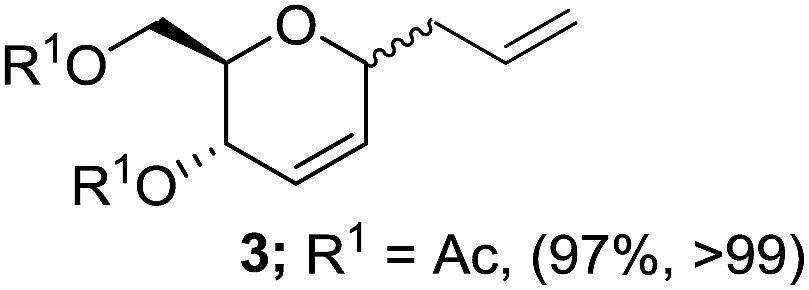 |
| 2 | 2a | 2 h | 4; R1 = Bn, (95%, α-only) |
| 3 | Me3SiCN, 2b | 1 h |  |
| 4 | 2b | 2 h | 6; R1 = Bn, (93%, 65:35) |
| 5 | Et3SiH, 2c | 2 h |  |
| 6 | Me3SiN3, 2d | 3 h |  |
| 7 | 2d | 6 h | 9; R1 = Bn, (15%, α-only), 9′; (72%, 67:33) |
| 8 | 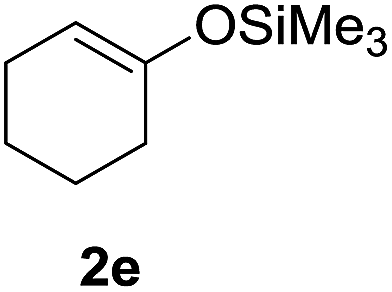 |
4 h |  |
| 9 |  |
3 h |  |
| 10 |  |
1 h | 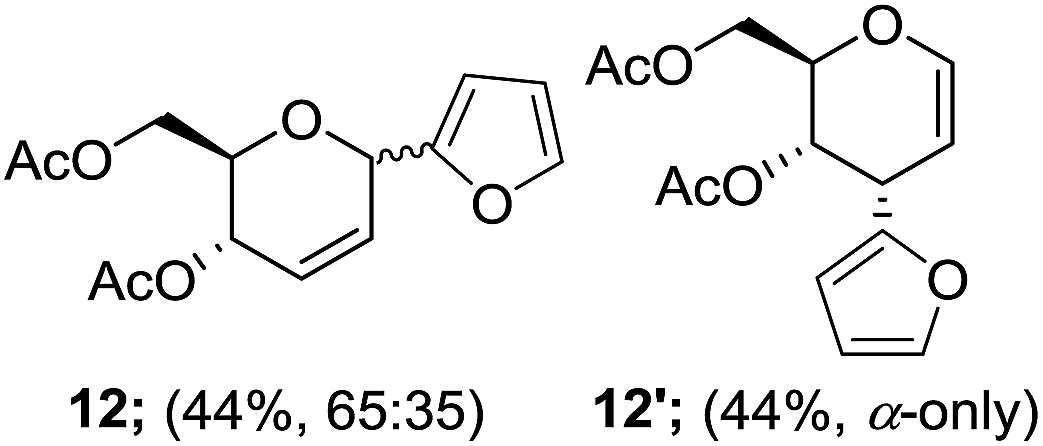 |
| 11 | 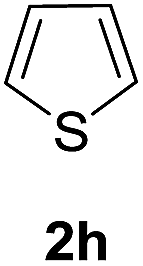 |
3 h |  |
We next focused on glycosylation of trimethylsilyl cyanide (2b) and trimethylsilyl azide (2d) to access the glycosyl cyanides and glycosyl azide since the glycosides containing azide and cyanides at anomeric center serve as useful chiral intermediates in the synthesis of various functionalized glycoconjugates.14
Thus, the reaction of 1a with 2b and triethylsilane (2c) proceeded smoothly and efficiently to give the corresponding glycosides (5, 7) in good yields (Table 1, entries 3 and 5). On the other hand, the C-glycosylation of trimethylsilyl azide (2d) produced the corresponding glycosyl azides as an inseparable mixture of C-1 (8) and C-3 (8′) epimers (entry 6). We next examined whether modification of protecting group in glycal impacted the stereochemistry of the products. Thus, we attempted the activation of 3,4,6-tri-O-benzyl glucal (1b) with 2a, 2b and 2d respectively under Zn(OTf)2-promoted glycosylation to obtain the desired glycosides (4, 6, 9). The outcome of these experiments revealed the formation of exclusive α-anomer when allyltrimethylsilane (2a) used as nucleophile. In contrast, the TMSCN (2b) and TMSN3 (2d) were reacted with 1b respectively to give the corresponding glucosides as inseparable mixture of isomers (entries 6 and 9). Notably, the glycal donor with acetyl leaving group at C-3 position reacted faster as compared to the corresponding benzyl ether protected glycal (entry 2, 4 and 7).
Furthermore, the silyl enol ether 2e and β-keto ester such as ethyl acetoacetate (2f) were successfully incorporated into glycal to obtain the desired C-glycosides (10–11) as stereoisomeric mixture due to prochirality at α-carbon (Table 1, entries 8 and 9). Due to the growing importance of sugar-heterocycles hybrid C-glycosides as key intermediates in the preparation of several biologically active molecules of medicinal significances,15 we next examined the glycosylation of heterocycles such as furan (2g) and thiophene (2h). Accordingly, the coupling of glycal 1a with 2g in the present reagent system resulted the mixture (1:1) of the corresponding C-3 (12′) and C-1 (12) epimers in 88% overall yields (entry 10). The spectroscopic analyses and consistency of the chemical shifts and distinctive coupling constants between the adjacent sugar protons with the literature data13c verified the α-configuration of C-3 epimer. On the other hand, the C-glycosylation of 2h with 1a under similar condition afforded the corresponding C-1 glycosyl heteroaromatic (13) in 74% yields as 1:1 α/β mixture while C-3 epimer was not observed (entry 11).
A plausible pathway of the regioisomers formation may be proposed based on analogy to previous reports on Ferrier reaction8c and Claisen rearrangement, a hetero [3,3]-sigmatropic rearrangement (Fig. 1).16 Accordingly, the regioselective formation of C-3-glycal (12′) with inversion of configuration indicates the potential anchimeric assistance by trans-oriented C-4 neighbouring acetyl group in half-chair twisted pyranoid conformation of oxocarbenium intermediate. Then, predominant formation of C-3 α-epimer depends on equilibration of the kinetic (oxocarbenium ion, B) to thermodynamic intermediate (A) under Lewis acid catalyzed glycosylation.
 | ||
| Fig. 1 Neighbouring group participation of C-4 substituent and hetero [3,3]-sigmatropic rearrangement. | ||
Eventually, the kinetically favoured intermediated (B) likely to rearranged into thermodynamically stable intermediate (A) to produced C-3 epimer as a major product. Unlike furan C-glycoside (12), glycosyl azide (8 and 9), an allylic azide undergoes a rapid sigmatropic rearrangement through a six-centered transition state (C) which results in dynamic equilibrium of all possible [3,3]-isomers. The predominant formation of C-3 epimers and sigmatropic rearrangements between C-1 to C-3 were observed only in case of glycosyl azide and furan sugars.
The scope of the reaction was further illustrated with other glycals. Accordingly, the coupling of 3,4,6-tri-O-acetyl D-galactal (1c) and acceptors 2a–2d were performed respectively under Zn-mediated C-glycosylation to obtain the desired galactosides (14–17) with excellent anomeric selectivity (Table 2, entries 1–4).
|
|||||
|---|---|---|---|---|---|
| Entry | Glycal | Acceptor | Time | Glycosideb; yield | α:β ratioc |
| a Reaction condition; glycal (1.0 equiv.), acceptor (1.2 equiv.), Zn(OTf)2 (10 mol%), 1,2-dichloroethane (2 mL), reaction temperature 40 °C.b Isolated and unoptimized yields.c The ratio were examined by spectroscopic analyses. | |||||
| 1 | 1c |  |
1 h | 14; 96% | α-only |
| 2 | 1c | 2b, Me3SiCN | 2 h | 15; 97% | α-only |
| 3 | 1c | 2c, Et3SiH | 3 h | 16; 98% | |
| 4 | 1c | 2d, Me3SiN3 | 6 h | 17; C-1, 25% | 93:07 |
| 17; C-3, 70% | 90:10 |
||||
| 5 | 1c |  |
6 h | 18; 93% | α-only |
| 6 | 1c |  |
3 h | 19; 78% | 55:45 |
| 7 | 1d |  |
2 h | 20; 80% | 93:07 |
| 8 | 1d | 2b, Me3SiCN | 1 h | 21; 72% | 60:40 |
| 9 | 1e (1f) |  |
1 h | 22; 91% (88%) | 88:12 |
| 10 | 1e (1f) | 2b, Me3SiCN | 1 h | 23; 93% (92%) | 85:15 |
| 11 | 1e (1f) | 2c, Et3SiH | 2 h | 24; 89% (87%) | |
| 12 | 1g |  |
3 h | 25; 95% | >99 |
| 13 | 1g | 2b, Me3SiCN | 2 h | 26; 84% | 68:32 |
| 14 | 1g | 2c, Et3SiH | 4 h | 27; 86% | |

Similarly, the reaction of sily enol ether (2e) proceeded in highly stereoselective manner to furnish the α-galactoside 18 in good yields (entry 5). On the other hand, the glycosylation reaction of thiophene (2h) with 1c resulted anomeric mixture and the C-linked thiophene galactoside (19) was isolated in 78% yield with ∼1:1 α/β mixture (entry 6).
We next probe the generality of reaction with deoxysugars such as rhamnal, xylal and arabinal. Therefore, the 3,4-di-O-acetyl-L-rhamnal (1d) was activated in the presence of silylated acceptors 2a–2b respectively to generate the corresponding C(2)–C(3) unsaturated glycosides (20–21) in good yields (Table 2, entries 7–8). Likewise, the glycosylation reaction of D-xylal (1e) and D-arabinal (1f) with 2a–2c were performed respectively to furnish the desired C-glycosides (22–24) with good yields and high anomeric selectivity (entries 9–11). The synthetic utility of the present method was further extended to incorporate the C-glycoside linkages in a disaccharide. Accordingly, the readily prepared peracetylated D-lactal (1g) was considered for Ferrier glycosylation with nucleophiles 2a–2c (entries 12–14). Remarkably, all the reactions proceeded efficiently and clean under mild reaction conditions and amenable the use of a disaccharide donor 1g to generate the desired C-linked disaccharides (25–27) containing various functionalities at anomeric position.
Encouraged by these results, we next envisioned a straight forward route for C-alkynylated sugar from glycals using activated alkyne since these chiral sugar derivatives could be transformed into various glycoconjugates.17 For this purpose, the phenyl (trimethylsilyl)acetylene (2i) was allowed to react with glucal 1a in the presence of 10 mol% Zn(OTf)2 in 1,2-dichloroethane. Notably, the reaction was completed in 2 h and afforded the desired C-1 alkynylated glucoside (28) in 97% yield with high α-anomeric selectivity (>99) (Scheme 1, entry 28). The glycosylation reaction of galactal (1c) with 2i afforded the corresponding galactoside (29) with exclusive α-anomer, which could be contributed by conformational equilibrium between 4H5 ↔ 5H4 and a vinylogous anomeric effect.18 Moreover, other glycals such as rhamnal (1d), xylal (1e), arabinal (1f) and lactal (1g) were subjected to C-alkynyl glycosylation with 2i respectively under similar conditions to afford the corresponding acetylene glycosides (30–32) in good yields and high anomeric selectivity (Scheme 1).‡
 | ||
| Scheme 1 Stereoselective synthesis of C-alkynylated glycosides. | ||
Having established the C-glycosylation of various glycals with silylated and heterocycles nucleophile, we extended our study to evaluate the reactivity of different glycals in C-alkylation by using trimethyl aluminium (2j) as model nucleophile. Thus, a solution of AlMe3 (2.0 M in hexane, 1.5 equiv.) in C2H4Cl2 (1 mL) was treated with Zn(OTf)2 (10 mol%) at 0 °C for 10 min under the atmosphere of argon. A preformed solution of glycal 1a in C2H4Cl2 (1 mL) was added dropwise to the above-mentioned mixture and reaction was allowed to stirred at 0 °C to room temperature. Remarkably, the complete conversion of starting material (glycal) was observed in 1 h to afford the desired C-methyl glucoside (33) in 94% yield with 80:20, α:β mixture (Table 3, entry 1).
|
||||
|---|---|---|---|---|
| Entry | Glycal donor | Glycoside | Condition A yieldb (α:β ratio)c |
Condition B19 yield (α:β ratio) |
| a For reaction condition, see general experimental procedure.b Isolated and unoptimized yields.c The anomeric ratios were examined by 1H NMR spectroscopy. | ||||
| 1 | D-Glucal (1a) | 33 | 94% (80:20) |
94% (79:21) |
| 2 | D-Galactal (1c) | 34 | 90% (>99) | 87% (89:11) |
| 3 | D-Xylal (1e) | 35 | 96% (88:12) |
71% (85:15) |
| 4 | D-Arbinal (1f) | 35 | 88% (88:12) |
81% (85:15) |
| 5 | D-Lactal (1g) | 36 | 92% (80:20) |
NA |

Subsequently, the generality of the Zn-mediated C-alkylation reaction was further evaluated in the glycosylation of AlMe3 (2j) with different glycals, the results and stereochemical outcomes are presented in Table 3. It is pertinent to mention that the C-alkylation of galactal (1c) with AlMe3 proceeded in highly stereoselective manner to afford the corresponding C-1 methyl 2,3-unsaturated galactoside (34) with exclusive α-anomeric selectivity (entry 2). In contrast, the coupling reaction of AlMe3 with deoxysugar xylal (1e) and arabinal (1f) furnished the desired methyl glycosides (35) in good yields with inseparable anomeric mixture (entries 3 and 4). Remarkably, the disaccharide glycal (1g) reacted smoothly under present mild reaction to afford the corresponding C-disaccharide (36) in 94% yields (entry 5).§ In addition, a distinctive comparison with the previous report19 on C-alkyl glycosylation further highlights the superiority of the Zn(OTf)2-mediated C-glycosylation in terms of reaction time, yields as well anomeric selectivity (Table 3).
In conclusion, we have reported a highly efficient and mild reagent system for incorporating C-glycosidic linkages in C(2)–C(3) unsaturated glycopyranosides. The scope and synthetic utility of the Zn-mediated protocol is further highlighted in the activation of different glycals with diverse range of acceptors to generate various functionalized glycosides. The present protocol is mild enough and amenable the use of a disaccharide glycal as well. We believed that this method would be useful in assembling oligosaccharides and glycoconjugates containing C-1 linkages at sugar moiety and would find its utility in carbohydrate chemistry.
Acknowledgements
Financial support from Department of Science and Technology (GAP0397 and 0471), New Delhi is gratefully acknowledged. The authors are also grateful to the Director CSIR-IICT for providing necessary infrastructure.References and notes
- (a) M. H. D. Postema, Tetrahedron, 1992, 48, 8545 CrossRef CAS; (b) Y. Du, R. J. Linhardt and I. R. Vlahov, Tetrahedron, 1998, 54, 9913 CrossRef CAS; (c) K. C. Nicolaou and H. J. Mitchell, Angew. Chem., 2001, 113, 1624 (Angew. Chem., Int. Ed., 2001, 40, 1576) CrossRef; (d) L. Somśak, Chem. Rev., 2001, 101, 81 CrossRef; (e) R. A. Dwek, Chem. Rev., 1996, 96, 683 CrossRef CAS PubMed; (f) G. V. M. Sharma and P. RadhaKrishna, Curr. Org. Chem., 2004, 8, 1187 CrossRef CAS; (g) A. Dondoni and A. Marra, Chem. Rev., 2000, 100, 4395 CrossRef CAS PubMed; (h) G. E. Ritchie, B. E. Moffatt, R. B. Sim, B. P. Morgan, R. A. Dwek and P. M. Rudd, Chem. Rev., 2002, 102, 305 CrossRef CAS PubMed; (i) S. Hanessian and B. Lou, Chem. Rev., 2000, 100, 4495 CrossRef.
- (a) M. D. Lewis, J. K. Cha and Y. Kishi, J. Am. Chem. Soc., 1982, 104, 4976 CrossRef CAS; (b) I. Paterson and L. E. Keown, Tetrahedron Lett., 1997, 38, 5727 CrossRef CAS; (c) K. Horita, Y. Sakurai, M. Nagasawa, S. Hachiya and O. Yonemitsu, Synlett, 1994, 43 CrossRef CAS; (d) B. Fraser-Reid, Acc. Chem. Res., 1985, 18, 347 CrossRef CAS; (e) M. M. Faul and B. E. Huff, Chem. Rev., 2000, 100, 2407 CrossRef CAS PubMed.
- (a) R. V. Weatherman, K. H. Mortell, M. Chervenak, L. L. Kiessling and E. J. Toone, Biochemistry, 1996, 35, 3619 CrossRef CAS PubMed; (b) D. P. Sutherlin, T. M. Stark, R. Hughes and R. W. Armstrong, J. Org. Chem., 1996, 61, 8350 CrossRef CAS PubMed; (c) W. Zou, Curr. Top. Med. Chem., 2005, 5, 1363 CrossRef CAS PubMed.
- T. Watanabe, G. Hira, M. Kato, D. Hashizume, T. Miyagi and M. Sodeka, Org. Lett., 2008, 10, 4167 CrossRef CAS PubMed.
- (a) R. J. Suhadolnik, Nucleoside Antibiotics, Wiley-Interscience, New York, 1970 Search PubMed; (b) J. Štambaský, M. Hocek and P. Kočovský, Chem. Rev., 2009, 109, 6729 CrossRef PubMed.
- (a) M. H. D. Postema, J. L. Piper, V. Komanduri and L. Liu, Angew. Chem., Int. Ed., 2004, 43, 2915 CrossRef CAS PubMed; (b) J. L. Piper and M. H. D. Postema, J. Org. Chem., 2004, 69, 7395 CrossRef CAS PubMed.
- (a) R. J. Ferrier, Chem. Soc., 1964, 5443 RSC; (b) D. M. Ciment and R. J. Ferrier, Chem. Soc. C, 1966, 441 RSC; (c) R. J. Ferrier and G. H. Sankey, J. Chem. Soc. C, 1966, 2345 RSC.
- (a) R. J. Ferrier and N. Prasad, J. Chem. Soc. C, 1969, 570 RSC; (b) R. J. Ferrier and N. Prasad, J. Chem. Soc. C, 1969, 581 RSC; (c) Review: A. M. Gómez, F. Lobo, C. Uriel and J. C. López, Eur. J. Org. Chem., 2013, 32, 7221 CrossRef; (d) A. A. Ansari, R. Lahiri and Y. D. Vankar, ARKIVOC, 2013, Pt. ii, 316 Search PubMed.
- (a) H. Y. Godage and A. J. Fairbanks, Tetrahedron Lett., 2000, 41, 7589 CrossRef CAS; (b) H. Kim, H. Men and C. Lee, J. Am. Chem. Soc., 2004, 126, 1336 CrossRef CAS PubMed; (c) J. Zeng, S. Xiang, S. Cai and X.-W. Liu, Angew. Chem., Int. Ed., 2013, 125, 1576 Search PubMed; (d) A. K. Kusunuru, M. Tatina, S. K. Yousuf and D. Mukherjee, Chem. Commun., 2013, 49, 10154 RSC; (e) M. Isobe, W. Phoosaha, R. Saeeng, K. Kira and C. Yenjai, Org. Lett., 2003, 5, 4883 CrossRef CAS PubMed; (f) A. S. Vieira, P. F. Fiorante, T. L. S. Hough, F. P. Ferreira, D. S. Lüdtke and H. A. Stefani, Org. Lett., 2008, 10, 5215 CrossRef CAS PubMed; (g) N. Lubin-Germain, A. H. Hallonet, F. Huguenot, S. Palmier, J. Uziel and J. Aüge, Org. Lett., 2007, 9, 3679 CrossRef CAS PubMed.
- (a) K. W. Wellington and S. A. Benner, Nucleosides, Nucleotides Nucleic Acids, 2006, 25, 1309 CrossRef CAS PubMed; (b) M. Lei, L. Gao and J.-S. Yang, Tetrahedron Lett., 2009, 50, 5135 CrossRef CAS; (c) H.-H. Li and X.-S. Ye, Org. Biomol. Chem., 2009, 7, 3855 RSC; (d) J. Ramnauth, O. Poulin, S. Rakhit and S. P. Maddaford, Org. Lett., 2001, 3, 2013 CrossRef CAS PubMed; (e) J. Ramnauth, O. Poulin, S. S. Bratovanov, S. Rakhit and S. P. Maddaford, Org. Lett., 2001, 3, 2571 CrossRef CAS PubMed; (f) S. Xiang, S. Cai, J. Zeng and X.-W. Liu, Org. Lett., 2011, 13, 4608 CrossRef CAS PubMed; (g) Y. Bai, L. M. H. Kim, H. Liao and X.-W. Liu, J. Org. Chem., 2013, 78, 8821 CrossRef CAS PubMed.
- (a) Regulatory guidelines for metal content; Doc. Ref. CPMP/SWP/QWP/4446/00, http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003588.pdf, accessed on 8 April 2014; (b) N. U. Kumar, B. S. Reddy, V. P. Reddy and R. Bandichhor, Tetrahedron Lett., 2012, 53, 4354 CrossRef.
- (a) G. Narasimha, B. Srinivas, P. RadhaKrishna and S. Kashyap, Synlett, 2014, 523 CAS; (b) B. Srinivas, G. Narasimha, P. RadhaKrishna and S. Kashyap, Synthesis, 2014, 1191 Search PubMed; (c) B. Srinivas, T. R. Reddy, P. RadhaKrishna and S. Kashyap, Synlett, 2014, 1325 Search PubMed; (d) S. Chittela, T. R. Reddy, P. RadhaKrishna and S. Kashyap, RSC Adv., 2014, 4, 46327 RSC; (e) T. R. Reddy, S. Chittela and S. Kashyap, Tetrahedron, 2014, 70, 9224 CrossRef CAS; (f) B. Srinivas, T. R. Reddy and S. Kashyap, Carbohydr. Res., 2015, 406, 86 CrossRef CAS PubMed; (g) S. K. Battina, T. R. Reddy, P. RadhaKrishna and S. Kashyap, Tetrahedron Lett., 2015, 56, 1798 CrossRef CAS; (h) T. R. Reddy, S. K. Battina and S. Kashyap, J. Carbohydr. Chem., 2015 DOI:10.1080/07328303.2015.1018994.
- (a) A. A. Ansari, Y. S. Reddy and Y. D. Vankar, Beilstein J. Org. Chem., 2014, 10, 300 CrossRef PubMed; (b) H. Kawabata, S. Kubo and M. Hayashi, Carbohydr. Res., 2001, 333, 153 CrossRef CAS PubMed; (c) J. S. Yadav, B. V. S. Reddy, J. V. Raman, N. Niranjan, S. K. Kumar and A. C. Kunwar, Tetrahedron Lett., 2002, 43, 2095 CrossRef CAS.
- (a) K. C. Nicolaou, M. E. Duggan, C. K. Hwang and P. K. Somers, J. Chem. Soc., Chem. Commun., 1985, 1359 RSC; (b) F. E. Wincott, S. J. Danishefsky and G. Schutte, Tetrahedron Lett., 1987, 28, 4951 CrossRef CAS; (c) S. Hanessian, Total Synthesis of Natural Products: The Chiron Approach, Pergamon Press, Oxford, 1984 Search PubMed; (d) D. E. Levy and C. Tan, The Chemistry of C-Glycosides, Pergamon, Oxford, 1995 Search PubMed.
- (a) R. R. Schmidt and G. Effenberger, Liebigs Ann. Chem., 1987, 825 CrossRef CAS; (b) M. J. Dianez, J. Galan, A. M. Gómez, J. C. López and M. Rico, J. Chem. Soc., Perkin Trans. 1, 1987, 581 RSC.
- (a) K. Heyns and R. Hohlweg, Chem. Ber., 1978, 111, 1632 CrossRef CAS; (b) A. K. Feldman, B. Colasson, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 13444 CrossRef CAS PubMed.
- M. Isobe, R. Nishizawa, S. Hosokawa and T. Nishizawa, Chem. Commun., 1998, 2665 RSC.
- (a) D. P. Curran and Y.-G. Suh, J. Am. Chem. Soc., 1984, 106, 5002 CrossRef CAS; (b) S. E. Denmark and M. S. Dappen, J. Org. Chem., 1984, 49, 798 CrossRef CAS; (c) A. R. Katritzky, P. J. Steel and S. N. Danisenko, Tetrahedron, 2001, 57, 3309 CrossRef CAS.
- P. Deelertpaiboon, V. Reutrakul, S. Jarussophon, P. Tuchinda, C. Kuhakarn and M. Pohmakotr, Tetrahedron Lett., 2009, 50, 6233 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General synthesis information and 1H NMR and 13C NMR spectra of all the glycosides are provided. See DOI: 10.1039/c5ra03328d |
| ‡ General experimental procedure for the synthesis of glycosides 3–32; to a stirred solution of glycal (1 equiv.) and acceptor (1.2 equiv.) in anhydrous 1,2-dichloroethane (2 mL mmol−1) under an atmosphere of argon was added Zn(OTf)2 (10 mol%) at room temperature. The reaction mixture was stirred at 40 °C until the complete consumption of the starting material (glycal), adjudged by TLC. The reaction was quenched with saturated NaHCO3 (5 mL), diluted with EtOAc (10 mL), and extracted with EtOAC (3 × 30 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2SO4, concentrated in vacuo and purified by silica gel column chromatography. All the compounds were confirmed by 1H NMR, 13C NMR and MS/HRMS spectroscopy and overall data were in complete agreement with the assigned structure. |
| § General experimental procedure for the synthesis of glycosides 33–36; to a stirred solution of Zn(OTf)2 (10 mol%) in 1,2-dichloroethane (2 mL mmol−1) in a two-necked round-bottom flask under an atmosphere of argon was added a solution of trimethylaluminum (2.0 M in hexanes, 1.5 equiv.) at 0 °C. The reaction mixture was stirred vigorously for 10–15 min at ice-cooled temperature. A preformed solution of glycal (1.0 equiv.) in anhydrous 1,2-dichloroethane (2 mL mmol−1) was added dropwise to the above-mentioned reaction mixture and reaction mixture was allowed to stirred at room temperature for 1 h. The reaction was quenched with saturated NaHCO3 (5 mL), diluted with EtOAc (10 mL), and extracted with EtOAC (3 × 30 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2SO4, concentrated in vacuo and purified by silica gel column chromatography to obtain the desired methyl glycosides. |
| This journal is © The Royal Society of Chemistry 2015 |