
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assigning regioisomeric or diastereoisomeric relations of problematic trisubstituted double-bonds through heteronuclear 2D selective J-resolved NMR spectroscopy†
Marie
Hoffmann
,
Solène
Miaskiewicz
,
Jean-Marc
Weibel
,
Patrick
Pale
* and
Aurélien
Blanc
*
Laboratoire de Synthèse, Réactivité Organiques et Catalyse, UMR 7177 associé au CNRS, Institut de Chimie, Université de Strasbourg, 4 rue Blaise Pascal, 67070 Strasbourg, France. E-mail: ablanc@unistra.fr; ppale@unistra.fr
First published on 15th April 2015
Abstract
Although one of the first 2D NMR methods, but so far neglected, selective J-resolved NMR spectroscopy offers a unique opportunity to help organic chemists in structure elucidation, avoiding natural and non-natural product misassignments. This NMR method indeed allowed us to unambiguously and simply assign (natural) product structures exhibiting trisubstituted unsaturations. For example, as demonstrated here, the isomeric aurone, flavone, coumarin and isocoumarin structures could easily be distinguished; regioisomer assignments in furans could be solved, as well as isomerisms in compounds containing trisubstituted double-bonds (aurones, lactones, divinyl ketones).
Introduction
In natural product research, structural and/or stereochemical assignments are obviously the key step, providing the foundation for a whole set of disciplines, from synthesis to biology, biochemistry or ecology, with often the production of new therapeutic agents as the most practical consequences. Despite the current sophisticated techniques available, assignments often prove delicate and sometimes cumbersome. The number of natural product structural misassignments reported each year is surprisingly high (Fig. 1).1 These numbers are probably underestimated, since those encountered misassignments are frequent and only revealed through total synthesis enterprises.2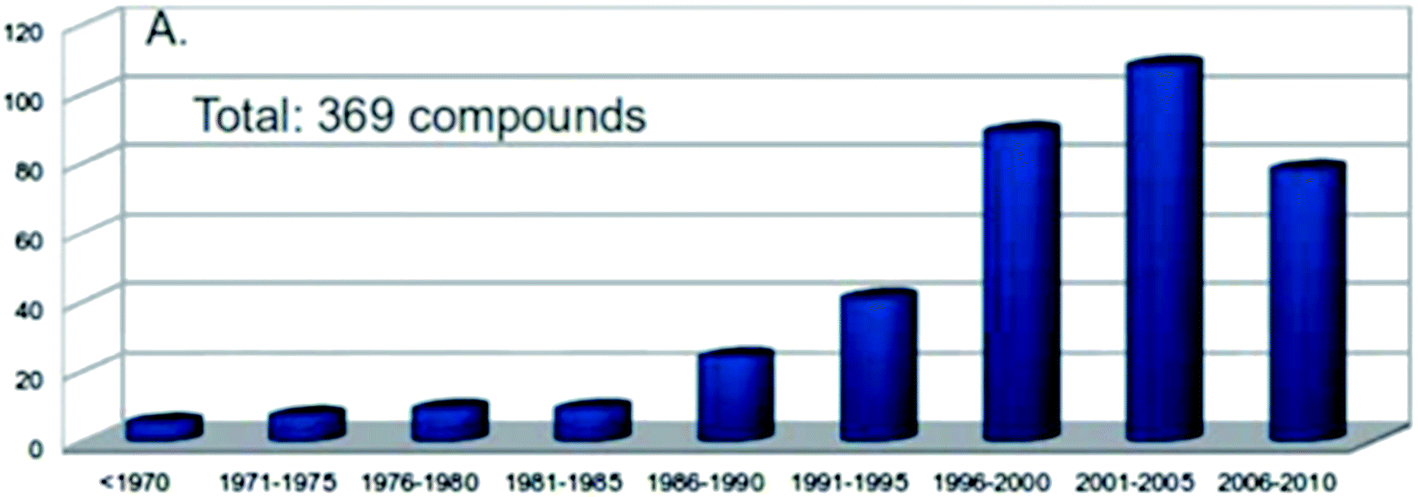 | ||
| Fig. 1 Numbers of known natural product misassignments per 5 year period. From ref. 1. | ||
Furthermore, the efforts and associated costs required to correct structures and appropriately reassign them could be quite significant and higher than those associated with the initial structure, especially in total synthesis endeavors.3 Besides the increased enrolments for total syntheses, wrong assignments might have dramatic effects due to the different biological activities the compounds could exhibit.
What was observed for natural products is also true in any synthetic efforts addressing compounds in which stereo- and/or regioisomers could be obtained, especially when only one is formed. In this area, the lack of re-synthesis minimizes the discovery of misassignments. Therefore, it is clear that, since the number of incorrectly determined structures of new products, natural or not, is quite large, reducing errors is clearly an important and useful challenge. Several routes are currently explored, among which are computer-assisted structure elucidation (CASE) method,4 NMR shift calculations combined with probability analyses5 or artificial neural network pattern recognition,6 combination of calculations with various spectroscopies, notably circular dichroism,7 and even atomic force microscopy imaging at atomic resolution.8 Despite these recent new developments, structure elucidation of new compounds, including natural products, heavily relies on two-dimensional NMR experiments. Correlation spectroscopy (COSY) and heteronuclear multiple and single bond correlation (HMBC/HSQC) detect interactions (couplings) of protons, respectively, with adjacent protons (from 2 to 4 bonds) or with neighbor carbon atoms (from 2 to 3 bonds). Both techniques provide connectivity between atoms. Stereochemistry and conformation of molecules are usually deduced from two-dimensional nuclear Overhauser enhancement spectroscopy (NOESY). Based on spin relaxation processes via dipolar coupling interacting through space, this method is able to detect protons that are in close proximity.
Facing various structural problems either in the total synthesis of natural products9 or in the development of new methods,10 we recently applied a neglected but simple NMR method, which successfully helped us to solve connectivity and/or stereochemistry problems. Being very efficient in our hands, we share here our results gained by applying the selective J-resolved NMR method during the process of structure elucidation with some typical examples.
Results and discussion
The (selective) J-resolved NMR method
Introduced in 1976 by Ernst et al.,11 the 2D J-resolved NMR experiments (JRES) are used to separate chemical shifts and J-couplings into two independent dimensions.12,13 The spin coupling may be homocoupling, usually between protons, or heterocoupling, essentially between 1H and 13C, although 1H and 31P have also been reported.14 The 2D spectra exhibit in one dimension the equivalent of a broad range decoupled proton or carbon spectrum and in the other the coupling, either 1H–1H or 13C–1H. Although one of the first 2D NMR experiments, JRES has been supplanted by the more recent COSY and HMBC methods in chemistry. These NMR techniques could also provide information about such coupling constants, but in a less practical way (see below).Interestingly, the JRES method is currently re-gaining interest for the study of complex mixtures,15 mostly in biology and medicine as a tool for metabolic screening16 and metabolomics17 as well as for biomarker identification, especially for cancer monitoring.18
Stereochemical double-bond assignment is still a problem for the trisubstituted ones,19 unfortunately quite common in natural products. Classically solved for disubstituted double-bonds with the E or Z3J coupling constants (1H NMR), the case of double-bonds carrying a single hydrogen is far from obvious. Coupling between vinylic and allylic hydrogens (4J) can be measured, but the corresponding E and Z values being low and close to each other do not allow unambiguous assignments.20 Due to the parallelism between 1H–1H and 13C–1H coupling constants, the E or Z geometry could also be reflected by the 3J or 2J (13C–1H) coupling constants.21 The usual way to measure them is to record 13C coupled 1H NMR.22,23 However, this technique leads to very complex spectra, due to overlapping of signals and to the complexity of multiplets, which may require computational method to solve out. Furthermore, rather long measurement times are needed and typically, signal-to-noise ratio is poor for such spectra.
Some of these aspects could also occur for the classic JRES method, but to a lower extent as all nJ (13C–1H) coupling constants are expressed in 2D. However, the heteronuclear selective J-resolved NMR experiment (SELJRES)12b elegantly solves this problem by inverting the resonance of the considered proton allowing to remove from the 2D map all other coupling constants excepted those of the selectively inverted proton:24 the required coupling can thus be easily available in one of the two dimensions.
Such stereochemical double-bond problems are often encountered in various families of natural products or in synthesis of unsaturated compounds, but only four typical examples will be detailed here.
Regioisomer assignment of flavonoids
Among flavonoids,25 coumarins, isocoumarins, aurones, and flavones constitute a sub-family contributing to the pigmentation of flowers and fruits, but also exhibiting interesting pharmaceutical activities.26 These four subclasses are structurally related, each one being isomeric to the others around a trisubstituted double-bond (Scheme 1). Due to their similarity, the structural elucidation of these natural products remains a challenge, and already led to numerous misassignments, from which only a few were detected and induced structural revision.9a,27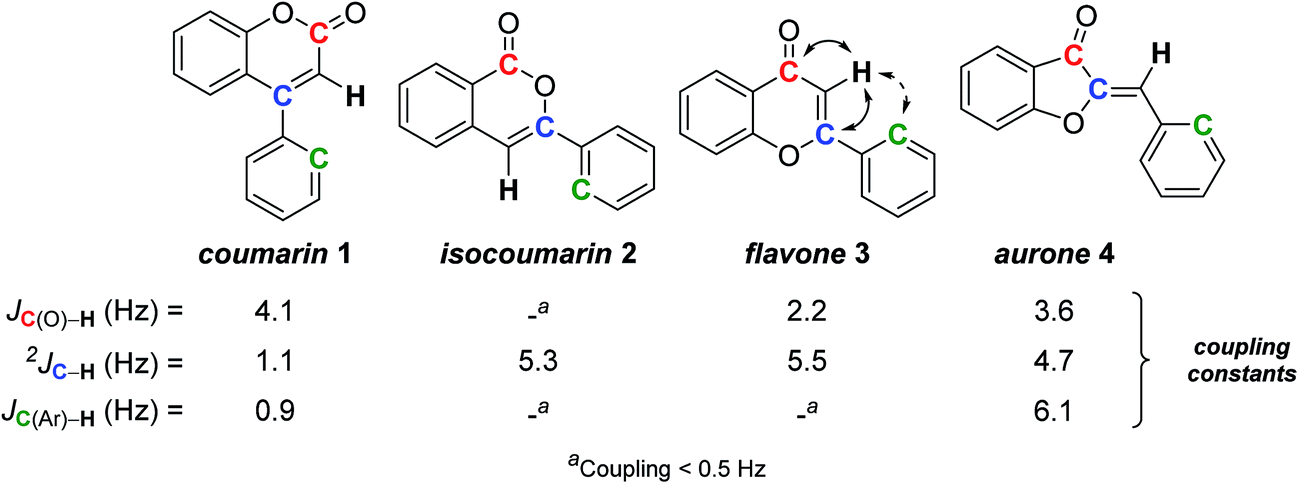 | ||
| Scheme 1 Relation structure-SELJRES coupling constants for flavonoid derivatives. | ||
Facing such problem, we – as others – relied on total synthesis of each possible isomer,9a but this gave us the opportunity to apply heteronuclear SELJRES and to find a useful and quick distinction between these isomers. Indeed, the couplings between the vinylic proton and the carbonyl carbon (red C in Scheme 1), the vinylic carbon (blue C) and the ortho carbon on the phenyl substituent (green C) in each series proved typical.28
Coumarin 1 showed a weak 2JCvinyl–H coupling constant of 1.1 Hz compared to the three other isomers 2–4 (2JCvinyl–H = 5.1 ± 0.4 Hz), probably reflecting the lack of oxygenated substituent on the double-bond. Isocoumarin 2 could easily be distinguished from the three other possible isomers by the lack of coupling between the vinylic proton and the carbonyl carbon. Flavone 3 and aurone 4 usually cannot be distinguished between themselves by their 1H NMR spectra due to a very similar shift of their vinylic proton (6.8–6.9 ppm) and other chemical shifts. However, they could be unambiguously distinguished by the presence or not of a coupling between the vinylic proton and the ortho carbon of the aromatic substituent: aurone 4 exhibits a 3J coupling (6.1 Hz) while flavone 3 can only have a 4J coupling, almost not detectable (Scheme 1).
Regioisomer assignment of trisubstituted furans
Furans constitute an important class of aromatic compounds, found in many natural products.29 Therefore, a large number of synthetic methods have been developed to build such five membered rings.30 Our group recently contributed to this area, achieving mild and convenient access to trisubstituted furans.31 During this study, as for furan natural product structural assignments, the regioselectivity has to be determined.With a single proton, such isomeric furans may be difficult to distinguish. When substituents in positions 2 and 5 do not carry hydrogen, there is no possibility to measure the 3J (1H–1H) coupling with the furanic proton. Since the situation of the furanic proton can be compared to those described in the previous section, we envisaged to solve this question by applying the SELJRES method. Indeed, the single proton should exhibit 2J coupling with the furanic carbon (red or blue C in Scheme 2), reflecting the proximity between the two atoms.
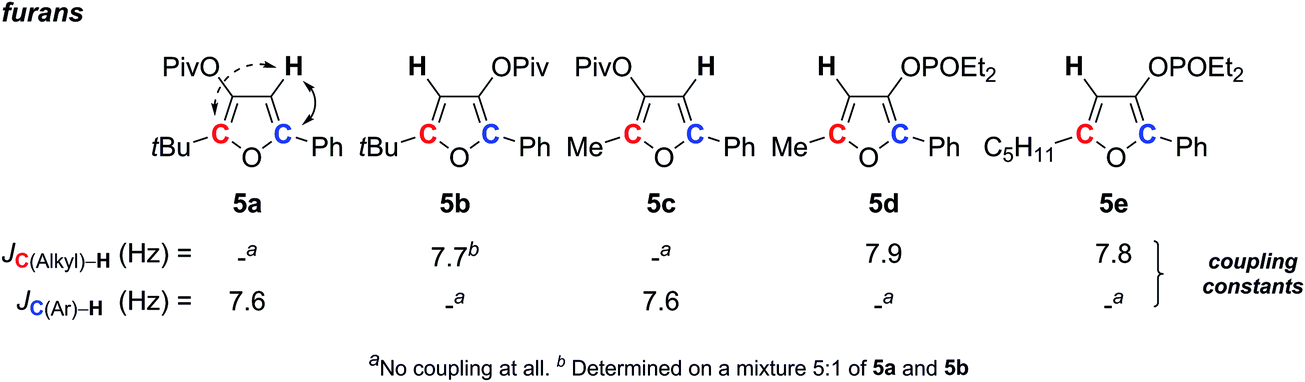 | ||
| Scheme 2 Relation structure-SELJRES coupling constants for furan derivatives. | ||
To look at this hypothesis, five different trisubstituted furans 5a–e were submitted to JRES by selecting the frequency of the furanic proton for acquisition. The analysis showed a 2J coupling constant around 7 to 8 Hz between the furanic proton and its adjacent β-carbon (red or blue C on Scheme 2). Interestingly, no 3J coupling was expressed with the γ-furanic carbon. This combination thus allowed for the unambiguous determination of the substitution pattern of furans. Furthermore, the analysis can be done on a mixture of two isomers as it was shown with 5a and 5b.
Diastereomer assignment of aurones and alkylidenelactones
As for the previous examples, the E or Z stereochemical assignment of trisubstituted double-bonds may not be obvious. This could also be true for methylenic systems. Aurones are typical of the former case, and our synthetic endeavor31 again provided us the opportunity to look at the JRES potential. Naturally occurring aurones usually exhibit a Z configuration of the double-bond, but such type of compounds can isomerize under irradiation or on silica gel.32 Alkylidene lactones also offer useful examples,9a,10b with the methylene lactones being typical of the latter case. Both allowed interesting comparisons.As before, SELJRES allowed us to get 2J or 3J (13C–1H) coupling constants, from which those between the vinylic proton and the carbonyl or vinylic carbons simply proved distinctive for each stereoisomer. The data collected for E or Z aurones 4 and 4a–d and related compound 4e clearly showed a correlation between such coupling constants and the stereochemistry of the double-bond (Scheme 3). The Z always exhibited 3J and/or 2J (13C–1H) values of 3–4 Hz, while the E isomer exhibited values of 7–8 Hz. Such clear-cut differences could obviously be fruitful in assigning stereochemistry and would have predictive value. This was confirmed with the alkylidene lactone series, the stereochemistry of which being already assigned by other techniques. Indeed, even in the simple methylene lactone 6, the same set of values was observed for the E and Z protons for the 3J (13C–1H) coupling (3.3 and 7.8 Hz, respectively) (Scheme 3). In other alkylidene lactones (Z)-7 and 8, similar trends could be observed with 3J (13C–1H) values of 3.5 ± 0.4 Hz noticed in Z isomers and 6.2 Hz for the E isomer (E)-7. In some specific cases, no 3J value was detected as for the bromomethylene furanone 9, structurally very close to the fimbrolide natural product family.33 However, a 2J coupling constant of 4.3 Hz was expressed in SELJRES, which could be attributed to the Z isomer by comparison with lactones (E)-7, (Z)-7 and 8.
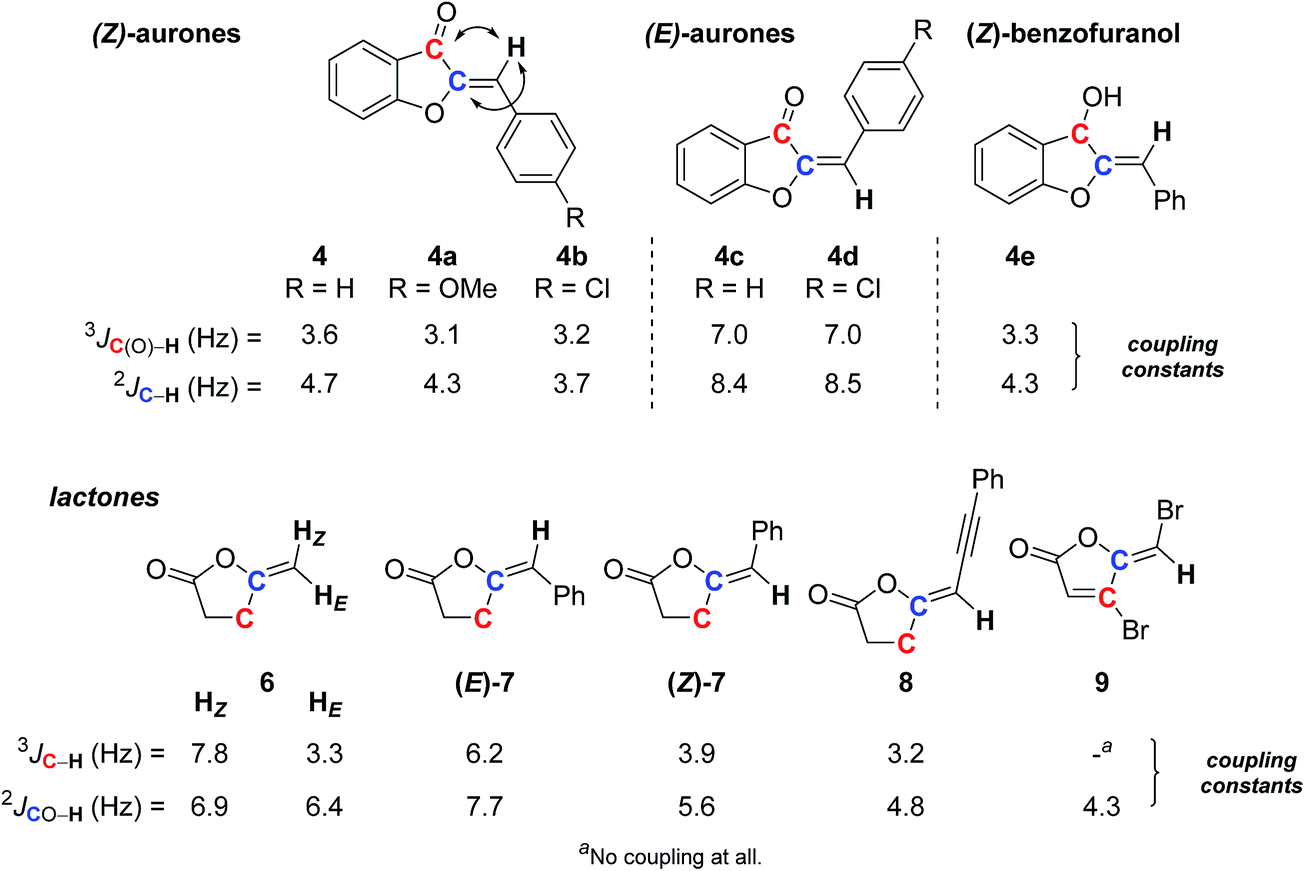 | ||
| Scheme 3 Simple E/Z assignment of aurones and lactone derivatives by SELJRES spectral analysis. | ||
It is worth mentioning that correlations from 1H-coupled 13C spectra with double-bond diastereoisomery have already been observed and compiled from around forty examples in the seventies (Table 1).21 The listed values are surprisingly large and similar to classical 1H–1H coupling constants, the E3J (13C–1H) coupling constants being always larger than the Z ones, but with a strong influence upon hybridization of the selected carbon. Although the general tendency is the same, with larger coupling constants for E relative to Z proton, the 3J SELJRES values we collected are far different from those already reported: between 3–4 Hz vs. 4–10 Hz for Z and 7–8 Hz vs. 9–17 Hz for E (Scheme 3vs.Table 1) and, in our study, no influence of the hybridization has been noticed (see compound 4vs.4e). More surprisingly, all our measured SELJRES values should correspond to cis proton/Z systems according to Table 1 data.
| C Hybridization | 3 J (C–H) coupling constant | ||
|---|---|---|---|
| H Z (Hz) | H E (Hz) | ||
|
sp (CN, CC) | 8.2–9.0 | 13.8–15.0 |
| sp2 (CO) | 4.3–10.0 | 9.5–16.9 | |
| sp3 (CH2X) | 5.7–8.0 | 7.7–11.0 | |
Diastereomer assignment of divinyl ketones
For a program devoted to gold catalysis and to the synthesis of cyclopentenone derivatives via the Nazarov reaction,34 we obtained various divinyl ketones,35 for which we had to assign the double-bond stereochemistry. Indeed, the reaction we developed led to divinyl ketones in which one double-bond stereochemistry was not defined by the synthetic pathway. In a first attempt, 2D NOESY was used, leading to some conclusions. However, based on the absence of correlation and due to the flexibility of such structures, assignments were suspicious. Fortunately, we were able to obtain crystal structures allowing us to unambiguously determine the stereochemistry. This helped us to look again at the correlations between 13C–1H SELJRES values and double-bond stereochemistry. Among the divinyl ketones we prepared, different examples were selected and submitted to SELJRES NMR experiments.A series of O-substituted divinyl ketones (10a–c and 11) was first evaluated, including one (10a) with X-ray crystallography structure (see ESI†) and one ((E)-10b and (Z)-10b) with both diastereomers. Their 3J and 2J (13C–1H) coupling constants, respectively, between the vinylic proton and the carbonyl carbon or the adjacent sp2 carbon were extracted (Scheme 4). Values similar to those already gained (see preceding section and Scheme 3) were obtained, with 3J and 2J (13C–1H) coupling constants of 8 ± 0.2 Hz for E isomers (10a and (E)-10b) and 3.6 ± 0.2 Hz for Z compounds ((Z)-10b, 10c and 11). These two sets of constants clearly depend on the geometry of the system and can easily be exploited to unambiguously assign double-bond geometry.
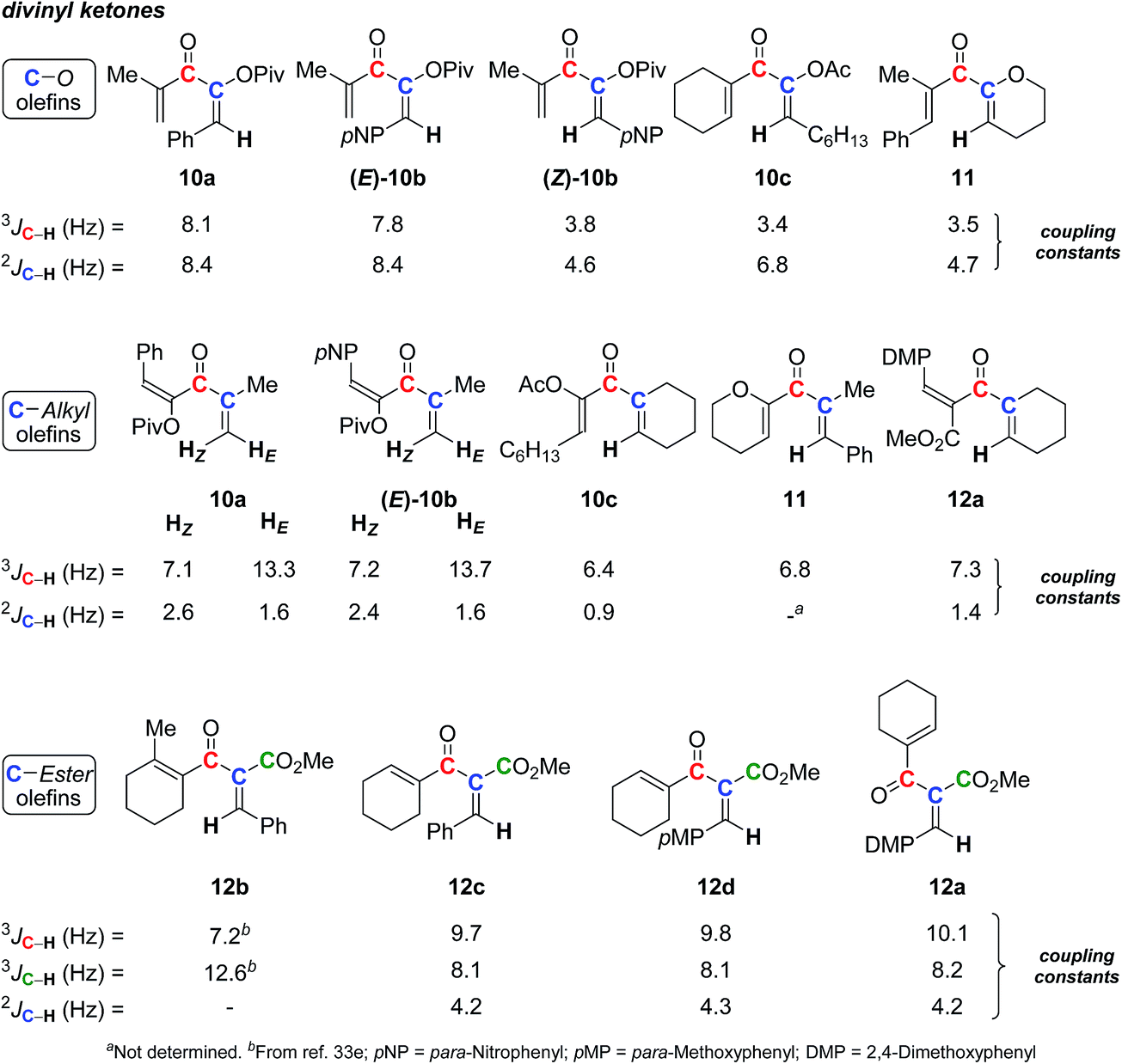 | ||
| Scheme 4 3 J and 2J (13C–1H) SELJRES values of variously substituted divinyl ketones 10–12. | ||
We also looked at the coupling constants of the double-bonds having C-substituents (10a–c, 11, and 12a). Once again, the geometry of each diastereomer could be easily determined by SELJRES with 3J (13C(O)–1H) values around 13 Hz for trans isomers (HE) and between 6.4 and 7.3 Hz for cis isomers (HZ). However, in the case of C-substituted divinyl ketones, the 2J (13C–1H) coupling constants did not follow the tendency of 3J (13C–1H) values: only weak values were measured (1.8 ± 0.8 Hz) without direct correlation with the E and Z nature of olefins.
Being well represented in the literature due to their good reactivity in the Nazarov reaction,36 divinyl ketones carrying an ester group (12a–d) were also investigated. In our case, only E isomers were available. SELJRES spectra showed that their 3J (13C(O)–1H) (trans values) and 3J (13CO2Me–1H) (cis values) coupling constants lay, respectively, between 9 to 10 Hz and around 8 Hz for compounds 12a and 12c–d. Working on the Nazarov reaction, Frontier et al. have also been interested in assigning the stereochemistry of divinyl ketones, and they have reported the 3J (13C–1H) extracted from 13C non-decoupled 1H NMR spectra, for Z and E isomers analogs.36e In this report, the coupling constants for E isomers correlated perfectly with the range of values obtained for 12a and 12c–d (9–10 Hz). However, the 3J (13CO–1H) (cis values, 4.6–7.2 Hz) and 3J (13CO2Me–1H) (trans values, 10.7–12.8 Hz) for Z isomers are far different, such as in 12b, which confirmed the Z nature of the considered olefin.
Conclusion
In this work, we have demonstrated that SELJRES NMR spectra gave direct and unambiguous access to the regioisomerism of various types of molecules, such as flavonoids and furans. SELJRES NMR thus offers a simple and easy method to rapidly determine the E and Z diastereochemistry of trisubstituted double-bonds in aurone, alkylidene lactone or divinyl ketone derivatives. Indeed, the 3J (13C–1H) coupling constants extracted from SELJRES are always larger for the E isomers, between 7–13 Hz, than for the Z isomers (3–8 Hz) depending on the substrate nature.Experimental section
General informations
Proton (1H NMR) and carbon (13C NMR) nuclear magnetic resonance spectra were recorded on a 500- or 600 MHz Bruker Avance II spectrometer equipped with a 5 mm DCH dual cryoprobe, with a z-gradient and operating at 500.130 MHz for 1H and 125.758 MHz for 13C. The solvent peak was used as reference value, for 1H NMR: CHCl3 = 7.26 ppm or benzene-d = 7.16 ppm and for 13C NMR: CHCl3 = 77.16 ppm or benzene-d = 128.06 ppm. For 1H NMR, data are presented as follows: chemical shift (ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, b = broad), coupling constants (J/Hz), integration, and attribution. For 13C NMR, data are presented as follows: chemical shift (ppm) and attribution. For 13C–{1H} selective J-resolved NMR, data are presented as follows: (chosen proton), 13C chemical shift (ppm) and coupling constant (J/Hz) or folded 1J (13C–1H), actually the acquisition method prevented the direct detection of the 1J (13C–1H) constant that is folded. Remark: despite all discussions about trisubstituted double-bonds, during the characterization it was observed that, for a chosen proton, sometimes the 2J (13C–1H) is smaller than its 3J (13C–1H).37 Assignments were determined either on the basis of unambiguous chemical shifts or coupling patterns, COSY, HSQC, HMBC, and NOESY experiments, to fully interprete spectra for related compounds. The pulse sequence used for measuring nJ (13C–1H) coupling constants is similar to the seljresqfsp sequence of the Bruker library. Experiments were acquired at 293 K with a relaxation delay of 3 s. Acquisition parameters were adjusted when necessary, but typically spectral windows were set to 25 kHz (200 ppm) for 13C and 50 Hz for nJ (13C–1H). Data size was at least 16k points in the direct dimension and 32 points in the indirect dimension. Broadband decoupling during acquisition was achieved by a composite pulse sequence Waltz16. Refocusing of 13C magnetization during evolution period was performed with an adiabatic shape pulse Crp60comp.4 of 2 ms. Simultaneously, a selective 180° pulse focused on the chosen proton frequency was achieved with a refocusing band-selective pulse (REBURP) of 80 ms. Melting points were recorded on a Melting Point Apparatus SMP3 and are uncorrected. Infrared spectra were recorded neat with Hewlett-Packard spectrometer. Wavelengths of maximum absorbance (νmax) are quoted in wave numbers (cm−1). High resolution mass spectra (HR-MS) data were recorded on a microTOF spectrometer equipped with orthogonal electrospray interface (ESI). The parent ions [M + Na]+ [M + H]+ is quoted. Analytical thin-layer chromatography (TLC) was carried out over silica gel 60 F254 plates or basic alumina (63–200 μm) with visualization by ultraviolet light, cerium ammonium molybdate (CAM) or potassium permanganate dip (KMnO4). Flash-column chromatography was carried out using silica gel 60 (40–63 μm) and the procedure included the subsequent evaporation of solvents in vacuo. Solvents were purified using standard means. 1,2-Dichloroethane (DCE), benzene and triethylamine (NEt3) were distilled from CaH2; pyridine (py) was distilled from potassium carbonate; diisopropylamine (DIPA) and N,N-diisopropylethylamine (DIPEA) were distilled from KOH; all under an argon atmosphere. Tetrahydrofuran (THF), acetonitrile (CH3CN) and dichloromethane (DCM) were dried using Glasstechnology GT S100 device. AuCl (Premion grade, 99.99%) and NaAuCl4·2H2O (Premion grade, 99.99%) were purchased from Alfa Aesar. AgSbF6 (98%) and PdCl2(dppf)·DCM were purchased from STREM Chemicals. IPrAuCl, PdCl2(PPh3)2 (98%), HNTf2 and CuI (98%) were purchased from Aldrich. AgNTf2 was prepared from HNTf2 (Aldrich) and Ag2CO3.38 All phosphinegold(I) chloride precatalysts were prepared by reduction of NaAuCl4·2H2O with thiodiethanol and subsequent addition of the appropriate phosphine.39 PPh3AuNTf2 catalyst was prepared from the corresponding triphenylphosphinegold(I) chloride and AgNTf2.40 All other chemicals were used as received.4-Phenyl-2H-chromen-2-one (1)
4-Phenyl-2H-chromen-2-one (1) was obtained in 91% yield over 2 steps from chromane-2,4-dione, using reported procedures (see ESI†).41 White solid; TLC Rf 0.23 (cyclohexane/EtOAc 20%); 1H NMR (500 MHz, CDCl3) δ 6.38 (s, 1H, H3), 7.23 (dd, J = 7.9, 7.1 Hz, 1H, H6), 7.41 (d, J = 8.1 Hz, 1H, H8), 7.44–7.47 (m, 2H, H2′), 7.50 (d, J = 7.9 Hz, 1H, H5), 7.52–7.55 (m, 3H, H3′/4′), 7.55 (dd, J = 8.1, 7.1 Hz, 1H, H7); 13C NMR (125.8 MHz, CDCl3) δ 115.3 (C3), 117.5 (C8), 119.1 (C4a), 124.3 (C6), 127.1 (C5), 128.6 (C2′), 129.0 (C3′), 129.8 (C4′), 132.1 (C7), 135.3 (C1′), 154.3 (C8a), 155.8 (C4), 160.9 (C2); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H3 6.38 ppm) δ 115.3 (folded 1JCH), 119.1 (3JCH = 7.2 Hz), 128.6 (4JCH = 0.9 Hz), 135.3 (3JCH = 5.0 Hz), 155.8 (2JCH = 1.1 Hz), 160.9 (2JCH = 4.1 Hz).3-Phenyl-1H-isochromen-1-one (2) (commercially available)
White solid; 1H NMR (500 MHz, CDCl3) δ 6.96 (s, 1H, H4), 7.41–7.53 (m, 5H, H3′/4′, H8 and H6), 7.72 (dd, J = 8.0, 6.9 Hz, 1H, H7), 7.89 (d, J = 8.1 Hz, 2H, H2′), 8.31 (d, J = 8.2 Hz, 1H, H5); 13C NMR (125.8 MHz, CDCl3) δ 102.0 (C4), 120.7 (C4a), 125.4 (C2′), 126.1 (C6), 128.3 (C8), 129.0 (C3′), 129.8 (C5), 130.1 (C4′), 132.1 (C1′), 135.0 (C7), 137.6 (C8a), 153.7 (C3), 162.5 (C1); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H4 6.96 ppm) δ 102.0 (folded 1JCH), 120.7 (2JCH = 5.4 Hz), 153.7 (2JCH = 5.3 Hz).2-Phenyl-4H-chromen-4-one (3)42
2-Phenyl-4H-chromen-4-one (3)42 was obtained in 41% yield over 3 steps from salicylaldehyde, using reported procedures (see ESI†).9a,43 White solid; TLC Rf 0.30 (cyclohexane/EtOAc 30%); 1H NMR (500 MHz, CDCl3) δ 6.83 (s, 1H, H3), 7.42 (dd, J = 8.1, 7.4 Hz, 1H, H6), 7.49–7.55 (m, 3H, H3′/4′), 7.57 (d, J = 8.1 Hz, 1H, H8), 7.70 (dd, J = 8.1, 7.4 Hz, 1H, H7), 7.93 (d, J = 7.7 Hz, 2H, H2′), 8.23 (d, J = 8.1 Hz, 1H, H5); 13C NMR (125.8 MHz, CDCl3) δ 107.7 (C3), 118.2 (C8), 124.1 (C4a), 125.4 (C6), 125.8 (C5), 126.4 (C2′), 129.2 (C3′), 131.7 (C4′), 131.9 (C1′), 133.9 (C7), 156.4 (C8a), 163.5 (C2), 178.6 (C4); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H3 6.83 ppm) δ 107.7 (folded 1JCH), 124.1 (3JCH = 3.8 Hz), 163.5 (2JCH = 5.5 Hz), 178.6 (2JCH = 2.2 Hz).Compounds 4, 4a, 4b and 4e were prepared from salicylaldehyde, using reported procedures (see ESI†).9a
(Z)-2-Benzylidenebenzofuran-3-one (4)9a
(Z)-2-Benzylidenebenzofuran-3-one (4)9a was obtained in 68% yield over 3 steps. Yellow solid; TLC Rf 0.16 (pentane/Et2O 10%); 1H NMR (500 MHz, CDCl3) δ 6.89 (s, 1H, H1′), 7.21 (dd, J = 7.8, 7.4 Hz, 1H, H5), 7.32 (d, J = 8.3 Hz, 1H, H7), 7.39 (t, J = 7.3 Hz, 1H, H5′), 7.45 (dd, J = 8.1, 7.3 Hz, 2H, H4′), 7.64 (dd, J = 8.3, 7.4 Hz, 1H, H6), 7.80 (d, J = 7.8 Hz, 1H, H4), 7.92 (d, J = 8.1 Hz, 2H, H3′); 13C NMR (125.8 MHz, CDCl3) δ 113.0 (C7), 113.1 (C1′), 121.7 (C3a), 123.6 (C5), 124.8 (C4), 129.0 (C4′), 129.995 (C5′), 131.6 (C3′), 132.4 (C2′), 137.0 (C6), 147.0 (C2), 166.2 (C7a), 184.9 (C3); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H1′ 6.89 ppm) δ 113.1 (folded 1JCH), 131.6 (3JCH = 6.1 Hz), 147.0 (2JCH = 4.7 Hz), 184.9 (3JCH = 3.6 Hz).(Z)-2-(4-Methoxybenzylidene)benzofuran-3-one (4a)9a
(Z)-2-(4-Methoxybenzylidene)benzofuran-3-one (4a)9a was obtained in 17% yield over 3 steps. Yellow solid; TLC Rf 0.38 (cyclohexane/EtOAc 30%); 1H NMR (500 MHz, CDCl3) δ 3.87 (s, 3H, Hp-OMe), 6.89 (s, 1H, H1′), 6.98 (d, J = 8.9 Hz, 2H, H4′), 7.21 (dd, J = 7.9, 7.2 Hz, 1H, H5), 7.32 (d, J = 8.3 Hz, 1H, H7), 7.64 (dd, J = 8.3, 7.2 Hz, 1H, H6), 7.80 (d, J = 7.9 Hz, 1H, H4), 7.92 (d, J = 8.9 Hz, 2H, H3′); 13C NMR (125.8 MHz, CDCl3) δ 55.5 (Cp-OMe), 113.0 (C7), 113.6 (C1′), 114.6 (C4′), 122.0 (C3a), 123.4 (C5), 124.7 (C4), 125.1 (C2′), 133.6 (C3′), 136.7 (C6), 146.0 (C2), 161.2 (C4′), 165.9 (C7a), 184.7 (C3); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H1′ 6.89 ppm) δ 113.6 (folded 1JCH), 133.6 (3J = 6.2 Hz), 146.0 (2JCH = 4.3 Hz), 184.7 (3JCH = 3.1 Hz).(Z)-2-(4-Chlorobenzylidene)benzofuran-3-one (4b)9a
(Z)-2-(4-Chlorobenzylidene)benzofuran-3-one (4b)9a was obtained in 39% yield over 3 steps. Yellow solid; TLC Rf 0.38 (cyclohexane/EtOAc 30%); 1H NMR (500 MHz, CDCl3) δ 6.82 (s, 1H, H1′), 7.23 (dd, J = 7.7, 7.0 Hz, 1H, H5), 7.33 (d, J = 8.4 Hz, 1H, H7), 7.42 (d, J = 8.9 Hz, 2H, H4′), 7.67 (dd, J = 8.4, 7.0 Hz, 1H, H6), 7.80 (d, J = 7.7 Hz, 1H, H4), 7.84 (d, J = 8.9 Hz, 2H, H3′); 13C NMR (125.8 MHz, CDCl3) δ 111.7 (C1′), 113.1 (C7), 121.6 (C3a), 123.8 (C5), 124.9 (C4), 129.3 (C4′), 130.9 (C5′), 132.7 (C3′), 136.0 (C2′), 137.2 (C6), 147.1 (C2), 166.193 (C7a), 184.8 (C3); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H1′ 6.82 ppm) δ 111.7 (folded 1JCH), 132.7 (3J = 6.1 Hz), 147.1 (2JCH = 3.7 Hz), 184.8 (3JCH = 3.2 Hz).(Z)-2-Benzylidene-2,3-dihydrobenzofuran-3-ol (4e)9a
(Z)-2-Benzylidene-2,3-dihydrobenzofuran-3-ol (4e)9a was obtained in 61% yield over 2 steps. White solid; TLC Rf 0.22 (cyclohexane/EtOAc 20%); 1H NMR (500 MHz, C6D6) δ 1.52 (br, 1H, HO), 5.32 (dd, J = 5.4, 1.6 Hz, 1H, H3), 5.83 (d, J = 1.6 Hz, 1H, H1′), 6.76 (dd, J = 7.6, 7.3 Hz, 1H, H5), 6.82 (d, J = 7.9 Hz, 1H, H7), 6.96 (dd, J = 7.9, 7.6 Hz, 1H, H6), 7.09 (t, J = 7.4 Hz, 1H, H5′), 7.18 (d, J = 7.3 Hz, 1H, H4), 7.28 (dd, J = 7.9, 7.4 Hz, 2H, H4′), 7.82 (d, J = 7.9 Hz, H3′); 13C NMR (125.8 MHz, CDCl3) δ 72.5 (C3), 106.1 (C1′), 110.6 (C7), 122.9 (C5), 125.9 (C4), 127.0 (C5′), 127.8 (C3a), 128.8 (C4′), 129.2 (C3′), 130.4 (C6), 135.4 (C2′), 157.6 (C2), 158.1 (C7a); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H1′ 5.83 ppm) δ 72.5 (3JCH = 3.3 Hz), 106.1 (folded 1JCH), 129.2 (3JCH = 5.8 Hz), 157.6 (2JCH = 4.3 Hz).(E)-2-Benzylidenebenzofuran-3-one (4c)
A solution of (Z)-2-benzylidenebenzofuran-3-one (4) (93 mg, 0.42 mmol) in CDCl3 (20 mL) was irradiated by the sunlight during 6 days. Solvent was then evaporated and the residue was purified by flash chromatography over silica gel (pentane/Et2O 5 to 10%) to yield the desired (E)-2-benzylidenebenzofuran-3-one in 42% yield (39 mg, 0.18 mmol). Starting material was also partially recovered (29%, 27 mg, 0.12 mmol). Yellow solid; mp 178 °C; TLC Rf 0.28 (pentane/Et2O 10%); IR (neat) νmax 481, 552, 598, 666, 696, 752, 857, 884, 1023, 1037, 1085, 1144, 1186, 1239, 1297, 1323, 1459, 1475, 1495, 1596, 1656, 1712, 1727 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.97 (s, 1H, H1′), 7.18 (dd, J = 8.2, 6.8 Hz, 1H, H5), 7.21 (d, J = 8.2 Hz, 1H, H7), 7.39–7.48 (m, 3H, H4′/5′), 7.63 (dd, J = 7.5, 6.8 Hz, 1H, H6), 7.79 (d, J = 7.5 Hz, 1H, H4), 8.17 (d, J = 7.9 Hz, 2H, H3′); 13C NMR (125.8 MHz, CDCl3) δ 112.7 (C7), 122.8 (C1′), 122.9 (C5), 123.5 (C3a), 124.8 (C4), 128.6 (C4′), 130.5 (C5′), 131.0 (C3′), 131.9 (C2′), 136.9 (C6), 148.2 (C2), 165.4 (C7a), 182.9 (C3); HR-MS 245.0606 (C15H10O2 + Na calcd 245.0573); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H1′ 6.97 ppm) δ 122.8 (folded 1JCH), 131.0 (3JCH = 6.3 Hz), 131.9 (2JCH = 1.7 Hz), 148.2 (2JCH = 8.4 Hz), 182.9 (3JCH = 7.0 Hz).(E)-2-(4-Chlorobenzylidene)benzofuran-3-one (4d)
(E)-2-(4-Chlorobenzylidene)benzofuran-3-one (4d) was obtained as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 mixture of 4b:4d over 1 step from the corresponding (Z) isomer (4b), after irradiation at 350 nm in methanol for 30 h.27a SELJRES NMR Analysis was directly performed on the crude mixture. 1H NMR (500 MHz, CDCl3) δ 7.25 (s, 1H, H1′), 7.53 (dd, J = 7.7, 7.6 Hz, 1H, H5), 7.55 (d, J = 8.2 Hz, 1H, H7), 7.74 (d, J = 8.6 Hz, 1H, H4′), 7.98 (dd, J = 8.2, 7.6 Hz, 1H, H6), 8.13 (d, J = 7.7 Hz, 1H, H4), 8.47 (d, J = 8.6 Hz, 2H, H3′); 13C NMR (125.8 MHz, CDCl3) δ 112.7 (C7), 121.4 (C1′), 123.0 (C5), 123.3 (C3a), 124.8 (C4), 128.8 (C4′), 130.4 (C2′), 132.3 (C3′), 136.3 (C5′), 137.1 (C6), 148.4 (C2), 165.4 (C7a), 183.0 (C3); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H1′ 7.25 ppm) δ 121.4 (folded 1JCH), 130.4 (2JCH = 1.6 Hz), 132.3 (3JCH = 6.4 Hz), 148.4 (2JCH = 8.5 Hz), 183.0 (3JCH = 7.0 Hz).
:6 mixture of 4b:4d over 1 step from the corresponding (Z) isomer (4b), after irradiation at 350 nm in methanol for 30 h.27a SELJRES NMR Analysis was directly performed on the crude mixture. 1H NMR (500 MHz, CDCl3) δ 7.25 (s, 1H, H1′), 7.53 (dd, J = 7.7, 7.6 Hz, 1H, H5), 7.55 (d, J = 8.2 Hz, 1H, H7), 7.74 (d, J = 8.6 Hz, 1H, H4′), 7.98 (dd, J = 8.2, 7.6 Hz, 1H, H6), 8.13 (d, J = 7.7 Hz, 1H, H4), 8.47 (d, J = 8.6 Hz, 2H, H3′); 13C NMR (125.8 MHz, CDCl3) δ 112.7 (C7), 121.4 (C1′), 123.0 (C5), 123.3 (C3a), 124.8 (C4), 128.8 (C4′), 130.4 (C2′), 132.3 (C3′), 136.3 (C5′), 137.1 (C6), 148.4 (C2), 165.4 (C7a), 183.0 (C3); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H1′ 7.25 ppm) δ 121.4 (folded 1JCH), 130.4 (2JCH = 1.6 Hz), 132.3 (3JCH = 6.4 Hz), 148.4 (2JCH = 8.5 Hz), 183.0 (3JCH = 7.0 Hz).
Compounds 5a, 5b and 5c were prepared using reported procedures (see ESI†).31
2-(tert-Butyl)-5-phenylfuran-3-yl pivalate (5a)31
2-(tert-Butyl)-5-phenylfuran-3-yl pivalate (5a)31 was obtained in 43% yield over 2 steps, from 4,4-dimethylpent-1-yn-3-yl pivalate. Colorless oil; TLC Rf 0.64 (pentane/Et2O 10%); 1H NMR (500 MHz, CDCl3) δ 1.35 (s, 9H, H3′′), 1.37 (s, 9H, H2′), 6.59 (s, 1H, H4), 7.23 (t, J = 7.3 Hz, 1H, H4′′′), 7.35 (dd, J = 8.0, 7.3 Hz, 2H, H3′′′), 7.59 (d, J = 8.0 Hz, 2H, H2′′′); 13C NMR (125.8 MHz, CDCl3) δ 27.3 (C3′′), 29.0 (C2′), 33.2 (C1′), 39.1 (C2′′), 102.7 (C4), 123.4 (C3′′′), 127.3 (C4′′′), 128.7 (C2′′′), 131.0 (C1′′′), 134.0 (C2), 148.9 (C5), 149.1 (C3), 176.4 (C1′′); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H4 6.59 ppm) δ 102.7 (folded 1JCH), 131.0 (3JCH = 1.0 Hz), 148.9 (2JCH = 7.6 Hz), 149.1 (2JCH = 4.9 Hz).5-(tert-Butyl)-2-phenylfuran-3-yl pivalate (5b)31
5-(tert-Butyl)-2-phenylfuran-3-yl pivalate (5b)31 was obtained in 5% yield over 2 steps, from 4,4-dimethylpent-1-yn-3-yl pivalate, and analyzed as a 5a:5b 5.1:1 mixture of regiosisomers. TLC Rf 0.71 (pentane/Et2O 10%); 1H NMR (500 MHz, CDCl3) δ 1.32 (s, 9H, H2′′′), 1.39 (s, 9H, H3′′), 6.24 (s, 1H, H4), 7.21 (t, J = 7.3 Hz, 1H, H4′), 7.38 (dd, J = 8.0, 7.3 Hz, 2H, H3′), 7.68 (d, J = 8.0 Hz, 2H, H2′); 13C NMR (125.8 MHz, CDCl3) δ 27.4 (C3′′), 29.0 (C2′′′), 33.1 (C1′′′), 39.3 (C2′′), 101.2 (C4), 123.9 (C3′), 126.7 (C4′), 128.6 (C2′), 130.3 (C1′), 135.7 (C2), 138.4 (C3), 161.6 (C5), 176.0 (C1′′); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H4 6.24 ppm) δ 101.2 (folded 1JCH), 138.4 (2JCH = 5.0 Hz), 161.6 (2JCH = 7.7 Hz).
2-Methyl-5-phenylfuran-3-yl pivalate (5c)44
2-Methyl-5-phenylfuran-3-yl pivalate (5c)44 was obtained in 22% yield over 2 steps, from but-3-yn-2-yl pivalate. White solid; TLC Rf 0.60 (pentane/Et2O 5%); 1H NMR (500 MHz, CDCl3) δ 1.35 (s, 9H, H3′′), 2.26 (s, 3H, H1′), 6.64 (s, 1H, H4), 7.23 (t, J = 7.3 Hz, 1H, H4′′′), 7.36 (dd, J = 7.8, 7.3 Hz, 2H, H3′′′), 7.59 (d, J = 7.8 Hz, 2H, H2′′′); 13C NMR (125.8 MHz, CDCl3) δ 10.8 (C1′), 27.3 (C3′′), 39.2 (C2′′), 102.1 (C4), 123.4 (C2′′′), 127.3 (C4′′′), 128.7 (C3′′′), 131.0 (C1′′′), 135.9 (C2), 139.8 (C3), 150.1 (C5), 176.2 (C1′′); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H4 6.64 ppm) δ 102.1 (folded 1JCH), 131.0 (3JCH = 1.1 Hz), 139.8 (2JCH = 5.4 Hz), 150.1 (2JCH = 7.6 Hz).Compounds 5d and 5e were prepared using reported procedures (see ESI†).31,45
Diethyl (5-methyl-2-phenylfuran-3-yl) phosphate (5d)45
Diethyl (5-methyl-2-phenylfuran-3-yl) phosphate (5d)45 was obtained in 10% yield over 5 steps, from but-1-yn-3-ol. Colorless oil; TLC Rf 0.30 (cyclohexane/EtOAc 20%); IR (neat) νmax 490, 578, 659, 693, 761, 800, 905, 1011, 1147, 1277, 1445, 1630, 2913, 2985 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.33 (td, J = 7.1 Hz, JHP = 0.9 Hz, 6H, H2′′), 2.32 (s, 3H, H2′′′), 4.15–4.27 (m, 4H, H1′′), 6.28 (s, 1H, H4), 7.21 (t, J = 7.5 Hz, 1H, H4′), 7.37 (dd, J = 8.6, 7.5 Hz, 2H, H3′), 7.73 (d, J = 8.6 Hz, 2H, H2′); 13C NMR (125.8 MHz, CDCl3) δ 14.3 (C2′′′), 16.2 (d, JCP = 6.9 Hz, C2′′), 64.9 (d, JCP = 6.4 Hz, C1′′), 103.5 (C4), 123.6 (C2′), 126.6 (C4′), 128.6 (C3′), 129.9 (C1′), 135.6 (d, JCP = 6.9 Hz, C2), 138.1 (d, JCP = 6.9 Hz, C3), 150.0 (C5); 31P NMR (162.0 MHz, CDCl3) δ −5.32; HR-MS 333.084 (C15H19O5P + Na calcd 333.086); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H4 6.28 ppm) δ 14.3 (3JCH = 1.2 Hz), 103.5 (folded 1JCH), 150.0 (2JCH = 7.9 Hz).Diethyl (5-pentyl-2-phenylfuran-3-yl) phosphate (5e)45
Diethyl (5-pentyl-2-phenylfuran-3-yl) phosphate (5e)45 was obtained in 24% yield over 5 steps, from oct-1-yn-2-ol (see ESI†). Colorless oil; TLC Rf 0.24 (cyclohexane/EtOAc 40%); 1H NMR (600 MHz, CDCl3) δ 0.90 (t, J = 7.1 Hz, 3H, H5′′′), 1.33 (td, J = 7.2 Hz, JHP = 1.1 Hz, 6H, H2′′), 1.33–1.37 (m, 4H, H4′′′/3′′′), 1.64–1.70 (m, 2H, H2′′′), 2.61 (t, J = 7.7 Hz, 2H, H1′′′), 4.16–4.26 (m, 4H, H1′′), 6.29 (s, 1H, H4), 7.20 (t, J = 7.4 Hz, 1H, H4′), 7.37 (dd, J = 8.6, 7.4 Hz, 2H, H3′), 7.73 (d, J = 8.6 Hz, 2H, H2′); 13C NMR (150.9 MHz, CDCl3) δ 14.1 (C5′′′), 16.2 (d, JCP = 6.8 Hz, C2′′), 22.5 (C4′′′), 27.5 (C2′′′), 28.6 (C1′′′), 31.4 (C3′′′), 64.9 (d, JCP = 6.1 Hz, C1′′), 102.7 (C4), 123.7 (C2′), 126.6 (C4′), 128.6 (C3′), 130.0 (C1′), 135.6 (d, JCP = 6.2 Hz, C2), 137.9 (d, JCP = 10.5 Hz, C3), 154.4 (C5); 31P NMR (121.5 MHz, CDCl3) δ −5.18; 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H4 6.29 ppm) δ 28.6 (3JCH = 1.0 Hz), 102.7 (folded 1JCH), 154.4 (2JCH = 7.8 Hz).Compounds 6, (Z)-7 and 8 were prepared using reported procedures (see ESI†).9b
5-Methylenedihydrofuran-2-one (6)9b
5-Methylenedihydrofuran-2-one (6)9b was obtained in 96% yield over 1 step, from pent-4-ynoic acid. Colorless oil; TLC Rf 0.18 (pentane/Et2O 10%); 1H NMR (500 MHz, CDCl3) δ 2.64–2.70 (m, 2H, H3), 2.85–2.91 (m, 2H, H4), 4.31 (dt, J = 2.2, 2.0 Hz, 1H, HE), 4.72 (td, J = 2.3, 2.2 Hz, 1H, HZ); 13C NMR (125.8 MHz, CDCl3) δ 25.2 (C4), 28.1 (C3), 88.9 (C1′), 155.7 (C5), 175.0 (C1); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: HZ 4.72 ppm) δ 25.2 (3JCH = 7.8 Hz), 28.1 (4JCH = 1.0 Hz), 88.9 (folded 1JCH), 155.7 (2JCH = 6.9 Hz); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: HE 4.31 ppm) δ 25.2 (3JCH = 3.3 Hz), 88.9 (folded 1JCH), 155.7 (2JCH = 6.4 Hz).(Z)-5-Benzylidenedihydrofuran-2-one ((Z)-7)9b
(Z)-5-Benzylidenedihydrofuran-2-one ((Z)-7)9b was obtained in 49% yield over 1 step, from 5-phenylpent-4-ynoic acid. Beige solid; TLC Rf 0.17 (cyclohexane/EtOAc 10%); 1H NMR (500 MHz, CDCl3) δ 2.68–2.74 (m, 2H, H3), 3.03 (td, J = 8.6, 1.8 Hz, 2H, H4), 5.55 (t, J = 1.8 Hz, 1H, H1′), 7.21 (t, J = 7.4 Hz, 1H, H5′), 7.33 (dd, J = 7.8, 7.4 Hz, 2H, H4′), 7.55 (d, J = 7.8 Hz, 2H, H3′); 13C NMR (125.8 MHz, CDCl3) δ 26.5 (C4), 27.1 (C3), 105.0 (C1′), 126.9 (C5′), 128.4 (C3′), 128.6 (C4′), 134.0 (C2′), 148.2 (C5), 175.1 (C1); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H1′ 5.55 ppm) δ 26.5 (3JCH = 3.9 Hz), 105.0 (folded 1JCH), 128.4 (3JCH = 5.6 Hz), 148.2 (2JCH = 5.6 Hz).(Z)-5-(3-Phenylprop-2-yn-1-ylidene)dihydrofuran-2-one (8)9b
(Z)-5-(3-Phenylprop-2-yn-1-ylidene)dihydrofuran-2-one (8)9b was obtained in 75% yield over 1 step, from 7-phenylheptan-4,6-diynoic acid. Yellow solid; TLC Rf 0.47 (cyclohexane/EtOAc 10%); 1H NMR (500 MHz, CDCl3) δ 2.72–2.77 (m, 2H, H3), 2.99 (td, J = 8.5, 1.9 Hz, 2H, H4), 5.04 (t, J = 1.9 Hz, 1H, H1′), 7.27–7.24 (m, 3H, H6′/7′), 7.42–7.48 (m, 2H, H5′); 13C NMR (125.8 MHz, CDCl3) δ 25.6 (C4), 27.5 (C3), 82.3 (C2′ or C3′), 85.6 (C1′), 93.8 (C2′ or C3′), 123.4 (C4′), 128.3 (C7′), 128.4 (C6′), 131.6 (C5′), 158.3 (C5), 173.9 (C1); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H1′ 5.04 ppm) δ 25.6 (3JCH = 3.2 Hz), 82.3 (2/3JCH = 2.3 Hz), 85.6 (folded 1JCH), 93.8 (2/3JCH = 4.6 Hz), 123.4 (4JCH = 1.7 Hz), 131.6 (5JCH = 1.7 Hz), 158.3 (2JCH = 4.8 Hz).(E)-5-Benzylidenedihydrofuran-2-one ((E)-7)
(E)-5-Benzylidenedihydrofuran-2-one ((E)-7) was obtained in 68% yield over 1 step from pent-4-ynoic acid, using reported procedure (see ESI†).46 Beige solid; TLC Rf 0.20 (pentane/Et2O 10%); 1H NMR (500 MHz, CDCl3) δ 2.73–2.79 (m, 2H, H3), 3.17 (td, J = 8.6, 2.2 Hz, 2H, H4), 6.33 (t, J = 2.2 Hz, 1H, H1′), 7.20–7.25 (m, 3H, H3′/5′), 7.35 (dd, J = 7.8, 7.8 Hz, 2H, H4′); 13C NMR (125.8 MHz, CDCl3) δ 25.3 (C4), 27.9 (C3), 107.2 (C1′), 126.8 (C5′), 127.9 (C3′), 128.8 (C4′), 134.5 (C2′), 151.3 (C5), 174.3 (C1); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H1′ 6.33 ppm) δ 25.3 (3JCH = 6.2 Hz), 107.2 (folded 1JCH), 127.9 (3JCH = 5.3 Hz), 151.3 (2JCH = 7.7 Hz).(Z)-4-Bromo-5-(bromomethylene)-2-furanone (9) (commercially available)
White solid; 1H NMR (500 MHz, CDCl3) δ 6.42 (s, 1H, H1′), 6.50 (s, 1H, H3); 13C NMR (125.8 MHz, CDCl3) δ 93.9 (C1′), 121.2 (C3), 135.4 (C5), 151.2 (C4), 165.6 (C1); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H1′ 6.42 ppm) δ 93.9 (folded 1JCH), 135.4 (2JCH = 4.3 Hz), 151.2 (3JCH = 10.6 Hz).Compounds 10a and (E)-10b were prepared using reported procedures (see ESI†).35a
(E)-4-Methyl-1-phenyl-2-pivaloxypenta-1,4-dien-3-one (10a)35a
(E)-4-Methyl-1-phenyl-2-pivaloxypenta-1,4-dien-3-one (10a)35a was obtained in 59% yield over 3 steps, from 2-methylbut-1-en-3-yne. Yellow solid; mp 35 °C; TLC Rf 0.51 (cyclohexane/EtOAc 20%); 1H NMR (500 MHz, CDCl3) δ 1.30 (s, 9H, H3′′), 1.82 (dd, J = 1.4, 1.2 Hz, 3H, H1′′′), 5.53 (dq, J = 1.6, 1.4 Hz, 1H, H5E), 5.94 (dq, J = 1.6, 1.2 Hz, 1H, H5Z), 6.84 (s, 1H, H1), 7.10–7.14 (m, 2H, H2′), 7.25–7.29 (m, 3H, H3′/4′); 13C NMR (125.8 MHz, CDCl3) δ 17.3 (C1′′′), 27.0 (C3′′), 38.7 (C2′′), 124.2 (C1), 127.8 (C5), 128.5 (C3′), 128.5 (C4′), 129.2 (C2′), 133.3 (C1′), 142.7 (C4), 144.0 (C2), 177.0 (C1′′), 193.3 (C3); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H1 6.84 ppm) δ 124.2 (folded 1JCH), 129.2 (3JCH = 6.4 Hz), 144.0 (2JCH = 8.4 Hz), 193.3 (3JCH = 8.1 Hz); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H5Z 5.94 ppm) δ 17.3 (3JCH = 10.3 Hz), 127.8 (folded 1JCH), 142.7 (2JCH = 2.6 Hz), 193.3 (3JCH = 7.1 Hz); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H5E 5.53 ppm) δ 17.3 (3JCH = 6.1 Hz), 127.8 (folded 1JCH), 142.7 (2JCH = 1.6 Hz), 193.3 (3JCH = 13.3 Hz).(E)-4-Methyl-1-(4-nitrophenyl)-2-pivaloxypenta-1,4-dien-3-one ((E)-10b)
(E)-4-Methyl-1-(4-nitrophenyl)-2-pivaloxypenta-1,4-dien-3-one ((E)-10b) was obtained in 18% yield over 3 steps, from 2-methylbut-1-en-3-yne (see ESI†). Yellowish solid; mp 78 °C; TLC Rf 0.42 (cyclohexane/EtOAc 20%); IR (neat) νmax 695, 738, 821, 858, 1037, 1103, 1152, 1202, 1273, 1342, 1513, 1592, 1667, 1720, 1745, 2874, 2935, 2976, 3079 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.29 (s, 9H, H3′′), 1.84 (dd, J = 1.7, 1.0 Hz, 3H, H1′′′), 5.67 (dq, J = 1.7, 1.2 Hz, 1H, H5E), 5.94 (dq, J = 1.2, 1.0 Hz, 1H, H5Z), 6.80 (s, 1H, H1), 7.30 (d, J = 8.9 Hz, 2H, H2′), 8.14 (d, J = 8.9 Hz, 2H, H3′); 13C NMR (125.8 MHz, CDCl3) δ 17.2 (C1′′′), 26.9 (C3′′), 38.7 (C2′′), 121.2 (C1), 123.8 (C3′), 128.3 (C5), 129.8 (C2′), 139.8 (C4′), 142.9 (C4), 146.5 (C2), 147.4 (C1′), 176.5 (C1′′), 192.1 (C3); HR-MS 340.1175 (C17H19NO5 + Na calcd 340.1155); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H1 6.80 ppm) δ 121.2 (folded 1JCH), 129.8 (3JCH = 4.7 Hz), 146.5 (2JCH = 8.4 Hz), 147.4 (2JCH = 1.0 Hz), 192.1 (3JCH = 7.8 Hz); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H5Z 5.94 ppm) δ 17.2 (3JCH = 10.2 Hz), 128.3 (folded 1JCH), 142.9 (2JCH = 2.4 Hz), 192.1 (3JCH = 7.2 Hz); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H5E 5.67 ppm) δ 17.2 (3JCH = 6.0 Hz), 128.3 (folded 1JCH), 142.9 (2JCH = 1.6 Hz), 192.1 (3JCH = 13.7 Hz).Compounds (Z)-10b and 10c were prepared using reported procedures (see ESI†).35a,47
(Z)-4-Methyl-1-(4-nitrophenyl)-2-pivaloxypenta-1,4-dien-3-one ((Z)-10b)
(Z)-4-Methyl-1-(4-nitrophenyl)-2-pivaloxypenta-1,4-dien-3-one ((Z)-10b) was obtained in 45% yield over 2 steps, from 2-methylbut-1-en-3-yne. Yellowish solid; mp 68 °C; TLC Rf 0.46 (pentane/Et2O 10%); IR (neat) νmax 691, 748, 778, 850, 892, 1031, 1092, 1164, 1270, 1340, 1514, 1596, 1656, 1716, 1751, 2874, 2934, 2977 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.35 (s, 9H, H3′′), 2.01 (dd, J = 1.4, 1.0 Hz, 3H, H1′′′), 5.86 (dq, J = 1.4, 1.2 Hz, 1H, H5), 5.90 (dq, J = 1.2, 1.0 Hz, 1H, H5), 6.88 (s, 1H, H1), 7.75 (d, J = 8.9 Hz, 2H, H2′), 8.23 (d, J = 8.9 Hz, 2H, H3′); 13C NMR (125.8 MHz, CDCl3) δ 18.4 (C1′′′), 27.0 (C3′′), 39.0 (C2′′), 123.7 (C1), 123.9 (C3′), 125.9 (C5), 130.7 (C2′), 138.9 (C4′), 142.6 (C4), 147.0 (C2), 147.7 (C1′), 176.0 (C1′′), 191.5 (C3); HR-MS 340.1175 (C17H19NO5 + Na calcd 340.1155); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H1 6.88 ppm) δ 123.7 (folded 1JCH), 130.7 (3JCH = 5.4 Hz), 147.0 (2JCH = 4.6 Hz), 191.5 (3JCH = 3.8 Hz).(Z)-2-Acetoxy-1-(cyclohex-1-enyl)non-2-en-1-one (10c)
(Z)-2-Acetoxy-1-(cyclohex-1-enyl)non-2-en-1-one (10c) was obtained in 90% yield over 2 steps, from ethynylcyclohex-1-ene. Colorless oil; TLC Rf 0.22 (pentane/Et2O 20%); IR (neat) νmax 590, 701, 745, 835, 883, 975, 1014, 1092, 1136, 1204, 1250, 1276, 1307, 1435, 1647, 1759, 2857, 2927 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.88 (t, J = 6.9 Hz, 3H, H9), 1.23–1.25 (m, 6H, H8/7/6), 1.43 (tt, J = 7.4, 7.1 Hz, 2H, H5), 1.59–1.69 (m, 4H, H4′′/5′′), 2.02 (dt, J = 7.6, 7.4 Hz, 2H, H4), 2.21 (s, 3H, H2′), 2.20–2.30 (m, 4H, H3′′/6′′), 6.05 (t, J = 7.6 Hz, 1H, H3), 6.69–6.73 (m, 1H, H2′′); 13C NMR (125.8 MHz, CDCl3) δ 14.2 (C9), 20.5 (C2′), 21.7 (C4′′), 22.0 (C5′′), 22.6 (C8), 24.0 (C6′′), 25.9 (C3′′), 26.1 (C4), 28.5 (C5), 29.1 (C6), 31.6 (C7), 131.9 (C3), 137.7 (C1′′), 140.8 (C2′′), 145.3 (C2), 169.0 (C1′), 191.1 (C1); HR-MS 301.175 (C17H26O3 + Na calcd 301.177); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H3 6.05 ppm) δ 26.1 (2JCH = 1.8 Hz), 28.5 (3JCH = 3.2 Hz), 131.9 (folded 1JCH), 145.3 (2JCH = 6.8 Hz), 191.1 (3JCH = 3.4 Hz); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H2′′ 6.69–6.73 ppm) δ 21.7 (3JCH = 5.5 Hz), 24.0 (3JCH = 7.2 Hz), 25.9 (2JCH = 3.7 Hz), 137.7 (2JCH = 0.9 Hz), 140.8 (folded 1JCH), 191.1 (3JCH = 6.4 Hz).(E)-1-(4,5-Dihydro-6H-pyran-2-yl)-2-methyl-3-phenylprop-2-en-1-one (11)48
(E)-1-(4,5-Dihydro-6H-pyran-2-yl)-2-methyl-3-phenylprop-2-en-1-one (11)48 was obtained in 26% yield over 2 steps from dihydropyrane, using reported procedure (see ESI†).35a Colorless oil; TLC Rf 0.40 (cyclohexane/EtOAc 30%); 1H NMR (500 MHz, CDCl3) δ 1.91 (tdd, J = 6.4, 6.0, 4.3 Hz, 2H, H5′), 2.13 (d, J = 1.4 Hz, 3H, H1′′), 2.26 (td, J = 6.4, 4.2 Hz, 2H, H4′), 4.17 (dd, J = 6.0, 4.3 Hz, 2H, H6′), 5.82 (t, J = 4.2 Hz, 1H, H3′), 7.24 (q, J = 1.4 Hz, 1H, H3), 7.29–7.36 (m, 1H, H4′′′), 7.35–7.42 (m, 4H, H2′′′/3′′′); 13C NMR (125.8 MHz, CDCl3) δ 14.9 (C1′′), 21.0 (C4′), 21.7 (C5′), 66.5 (C6′), 113.4 (C3′), 128.4 (C4′′′), 128.5 (C3′′′), 129.7 (C2′′′), 135.9 (C1′′′ or C2), 136.0 (C1′′′ or C2), 138.9 (C3), 151.4 (C2′), 193.9 (C1); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H3′ 5.82 ppm) δ 14.9 (3JCH = 8.3 Hz), 21.0 (2JCH = 2.6 Hz), 21.7 (3JCH = 5.5 Hz), 113.4 (folded 1JCH), 151.4 (2JCH = 4.7 Hz), 193.9 (3JCH = 3.5 Hz); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H3 7.24 ppm) δ 138.9 (folded 1JCH), 193.9 (3JCH = 6.8 Hz).Compounds 12a, 12c and 12d were prepared using reported procedures (see ESI†).36e,49
Methyl (E)-2-(cyclohex-1-enylcarbonyl)-3-(2,4-dimethoxyphenyl)prop-2-enoate (12a)
Methyl (E)-2-(cyclohex-1-enylcarbonyl)-3-(2,4-dimethoxyphenyl)prop-2-enoate (12a) was obtained in 64% yield over 2 steps, from cyclohex-1-en-1-carboxylic acid. Yellowish solid; mp 99 °C; TLC Rf 0.09 (pentane/Et2O 30%); IR (neat) νmax 401, 461, 530, 768, 802, 826, 927, 995, 1030, 1044, 1072, 1124, 1162, 1192, 1207, 1237, 1301, 1313, 1422, 1434, 1459, 1501, 1593, 1632, 1646, 1706, 2836, 2929, 2945 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.50–1.57 (m, 2H, H4′′′), 1.58–1.64 (m, 2H, H5′′′), 2.07–2.14 (m, 2H, H3′′′), 2.29–2.35 (m, 2H, H6′′′), 3.77 (s, 3H, HOMe), 3.79 (s, 3H, Ho- or p-OMe), 3.82 (s, 3H, Ho- or p-OMe), 6.35 (dd, J = 8.8, 2.5 Hz, 1H, H3′ or 5′), 6.39 (d, J = 2.5 Hz, 1H, H3′ or 5′), 6.80–6.84 (m, 1H, H2′′′), 7.12 (d, J = 8.8 Hz, 1H, H6′), 8.14 (s, 1H, H3); 13C NMR (125.8 MHz, CDCl3) δ 21.6 (C4′′′), 21.9 (C5′′′), 22.8 (C6′′′), 26.3 (C3′′′), 52.4 (COMe), 55.6 (Co- or p-OMe), 55.6 (Co- or p-OMe), 98.3 (C3′ or 5′), 105.0 (C3′ or 5′), 115.6 (C1′), 128.1 (C2), 131.2 (C6′), 136.9 (C3), 139.2 (C1′′′), 144.8 (C2′′′), 159.8 (C2′), 162.9 (C4′), 166.5 (C1), 197.8 (C1′′); HR-MS 353.1389 (C19H22O5 + Na calcd 353.1359); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H3 8.14 ppm) δ 128.1 (2JCH = 4.2 Hz), 131.2 (3JCH = 6.3 Hz), 136.9 (folded 1JCH), 159.8 (3JCH = 3.5 Hz), 166.5 (3JCH = 8.2 Hz), 197.8 (3JCH = 10.1 Hz); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H2′′′ 6.80–6.84 ppm) δ 21.6 (3JCH = 5.3 Hz), 22.8 (3JCH = 6.8 Hz), 26.3 (2JCH = 3.7 Hz), 139.2 (2JCH = 1.4 Hz), 144.8 (folded 1JCH), 197.8 (3JCH = 7.3 Hz).Methyl (E)-2-(cyclohex-1-enylcarbonyl)-3-phenylprop-2-enoate (12c)
Methyl (E)-2-(cyclohex-1-enylcarbonyl)-3-phenylprop-2-enoate (12c) was obtained in 54% yield over 2 steps, from cyclohex-1-en-1-carboxylic acid. Yellowish solid; mp 43 °C; TLC Rf 0.19 (cyclohexane/EtOAc 10%); IR (neat) νmax 401, 492, 551, 580, 680, 770, 830, 929, 992, 1068, 1102, 1185, 1212, 1391, 1421, 1497, 1619, 1647, 1713, 2861, 2929 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.52–1.58 (m, 2H, H4′′′), 1.60–1.66 (m, 2H, H5′′′), 2.09–2.14 (m, 2H, H3′′′), 2.32–2.38 (m, 2H, H6′′′), 3.79 (s, 3H, HOMe), 6.83 (t, J = 7.6 Hz, 1H, H2′′′), 7.29–7.35 (m, 5H, H2′/3′/4′), 7.81 (s, 1H, H3); 13C NMR (125.8 MHz, CDCl3) δ 21.5 (C4′′′ or C5′′′), 21.8 (C4′′′ or C5′′′), 22.8 (C6′′′), 26.4 (C3′′′), 52.7 (COMe), 128.8 (C3′), 130.1 (C2′), 130.3 (C4′), 131.4 (C2), 133.4 (C1′), 139.3 (C1′′′), 142.1 (C3), 145.6 (C2′′′), 165.9 (C1), 196.9 (C1′′); HR-MS 293.114 (C17H18O3 + Na calcd 293.115); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H3 7.81 ppm) δ 130.1 (3JCH = 5.6 Hz), 131.4 (2JCH = 4.2 Hz), 133.4 (2JCH = 1.2 Hz), 142.1 (folded 1JCH), 165.9 (3JCH = 8.1 Hz), 196.9 (3JCH = 9.7 Hz).Methyl (E)-2-(cyclohex-1-enylcarbonyl)-3-(4-methoxyphenyl)prop-2-enoate (12d)
Methyl (E)-2-(cyclohex-1-enylcarbonyl)-3-(4-methoxyphenyl)prop-2-enoate (12d) was obtained in 56% yield over 2 steps, from cyclohex-1-en-1-carboxylic acid. Colorless oil; TLC Rf 0.16 (pentane/Et2O 30%); IR (neat) νmax 520, 540, 830, 924, 989, 1027, 1070, 1119, 1171, 1200, 1250, 1305, 1433, 1511, 1599, 1632, 1651, 1704, 2839, 2859, 2933 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.54–1.61 (m, 2H, H4′′′), 1.63–1.69 (m, 2H, H5′′′), 2.11–2.16 (m, 2H, H3′′′), 2.35–2.40 (m, 2H, H6′′′), 3.78 (s, 3H, HOMe), 3.81 (s, 3H, Hp-OMe), 6.83 (d, J = 8.9 Hz, 2H, H3′), 6.85–6.88 (m, 1H, H2′′′), 7.27 (d, J = 8.9 Hz, 2H, H2′), 7.75 (s, 1H, H3); 13C NMR (125.8 MHz, CDCl3) δ 21.6 (C4′′′ or C5′′′), 21.8 (C4′′′ or C5′′′), 22.8 (C6′′′), 26.4 (C3′′′), 52.5 (COMe), 55.5 (Cp-OMe), 114.3 (C3′), 125.9 (C1′), 128.7 (C2), 132.2 (C2′), 139.3 (C1′′′), 141.8 (C3), 145.4 (C2′′′), 161.3 (C4′), 166.2 (C1), 197.5 (C1′′); HR-MS 323.125 (C18H20O4 + Na calcd 323.125); 13C–{1H} selective J-resolved NMR (125.8 MHz, CDCl3, chosen proton: H3 7.75 ppm) δ 125.9 (2JCH = 1.1 Hz), 128.7 (2JCH = 4.3 Hz), 132.2 (3JCH = 5.9 Hz), 141.8 (folded 1JCH), 166.2 (3JCH = 8.1 Hz), 197.5 (3JCH = 9.8 Hz).Acknowledgements
We gratefully acknowledge the Agence Nationale de la Recherche for a grant (ANR-11-JS07-001-01 Synt-Het-Au) and the CNRS. M. H. and S. M. thank the French Ministry of Research for a PhD fellowships. M. H. also thanks Dr L. Allouche and M. Coppe (Service de RMN, Faculté de Chimie, 1 rue Blaise Pascal, University of Strasbourg, Strasbourg, France) for their fruitful help in acquisition of SELJRES spectra.Notes and references
- T. L. Suyama, W. H. Gerwick and K. L. McPhail, Bioorg. Med. Chem., 2011, 19, 6675–6701 CrossRef CAS PubMed.
- K. C. Nicolaou and S. A. Snyder, Angew. Chem., Int. Ed., 2005, 44, 1012–1044 CrossRef CAS PubMed.
- For selected examples of natural product structure revisions, see: (a) K. Ogawa, Y. Koyama, I. Ohashi, I. Sato and M. Hirama, Angew. Chem., Int. Ed., 2009, 48, 1110–1113 CrossRef CAS PubMed; (b) S. Kobayashi, S. Ashizawa, Y. Takahashi, Y. Sugiura, M. Nagaoka, M. J. Lear and M. Hirama, J. Am. Chem. Soc., 2001, 123, 11294–11295 CrossRef CAS PubMed.
- M. Elyashberg, A. J. Williams and K. Blinov, Nat. Prod. Rep., 2010, 27, 1296–1328 RSC.
- S. G. Smith and J. M. Goodman, J. Am. Chem. Soc., 2010, 132, 12946–12959 CrossRef CAS PubMed.
- A. M. Sarotti, Org. Biomol. Chem., 2013, 11, 4847–4859 CAS.
- A. Bihlmeier, E. Bourcet, S. Arzt, T. Muller, S. Bräse and W. Klopper, J. Am. Chem. Soc., 2011, 134, 2154–2160 CrossRef PubMed.
- L. Gross, F. Mohn, N. Moll, G. Meyer, R. Ebel, W. M. Abdel-Mageed and M. Jaspars, Nat. Chem., 2010, 2, 821–825 CrossRef CAS PubMed.
- (a) H. Harkat, A. Blanc, J.-M. Weibel and P. Pale, J. Org. Chem., 2008, 73, 1620–1623 CrossRef CAS PubMed; (b) H. Harkat, A. Y. Dembelé, J.-M. Weibel, A. Blanc and P. Pale, Tetrahedron, 2009, 65, 1871–1879 CrossRef CAS.
- (a) H. Harkat, J.-M. Weibel and P. Pale, Tetrahedron Lett., 2006, 47, 6273–6276 CrossRef CAS; (b) H. Harkat, J.-M. Weibel and P. Pale, Tetrahedron Lett., 2007, 48, 1439–1442 CrossRef CAS; (c) A. Blanc, K. Tenbrink, J.-M. Weibel and P. Pale, J. Org. Chem., 2009, 74, 4360–4363 CrossRef CAS PubMed; (d) N. Kern, M. Hoffmann, A. Blanc, J.-M. Weibel and P. Pale, Org. Lett., 2013, 15, 836–839 CrossRef CAS PubMed.
- (a) W. P. Aue, E. Bartholdi and R. R. Ernst, J. Chem. Phys., 1976, 64, 2229–2246 CrossRef CAS; (b) G. Bodenhausen, R. Freeman and D. L. Turner, J. Magn. Reson., 1977, 27, 511–514 CAS.
- (a) M. Bruch, NMR Spectroscopy Techniques, CRC Press, 2nd edn, 1996 Search PubMed; (b) T. D. W. Claridge, Separating shifts and couplings: J-resolved spectroscopy, in High-resolution NMR Techniques in Organic Chemistry, Elsevier, Oxford, 2nd edn, 2009, 27, ch. 7, Tetrahedron Organic Chemistry Series, pp. 233–246 Search PubMed.
- For recent improvement in JRES NMR spectroscopy, see: A. Martinez, F. Bourdreux, E. Riguet and J.-M. Nuzillard, Magn. Reson. Chem., 2012, 50, 28–32 CrossRef CAS PubMed.
- E. Cesarotti, M. Grassi and L. Prati, J. Chem. Soc., Dalton Trans., 1989, 161–168 RSC.
- R. Novoa-Carballal, E. Fernandez-Megia, C. Jimenez and R. Riguera, Nat. Prod. Rep., 2010, 28, 78–98 RSC.
- J. M. Fonville, A. D. Maher, M. Coen, E. Holmes, J. C. Lindon and J. K. Nicholson, Anal. Chem., 2010, 82, 1811–1821 CrossRef CAS PubMed.
- C. Ludwig and M. R. Viant, Phytochem. Anal., 2010, 21, 22–32 CrossRef CAS PubMed.
- T. Lange, R. F. Schulte and P. Boesiger, Magn. Reson. Med., 2008, 59, 966–972 CrossRef PubMed.
- For a recent example, see: K. D. Klika, S. Cheriyamundath, R. Raghavan, K. B. Megha, A. Banerji, R. W. Owen and J. Madassery, Tetrahedron Lett., 2014, 55, 6550–6553 CrossRef CAS.
- R. M. Silverstein, F. X. Webster and D. Kiemle, Spectrometric Identification of Organic Compounds, Wiley Global Education, 7th edn, 2005 Search PubMed.
- (a) C. A. Kingsbury, D. Draney, A. Sopchik, W. Rissler and D. Durham, J. Org. Chem., 1976, 41, 3863–3868 CrossRef CAS; (b) F. H. A. Rummens and J. W. de Haan, Org. Magn. Reson., 1970, 2, 351–355 CrossRef CAS.
- For others methods to measure long range CH spin coupling constants, see: T. Parella and J. F. Espinosa, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 73, 17–55 CrossRef CAS PubMed.
- For examples of trisubstituted olefins assignment by 13C non-decoupled 1H NMR spectroscopy, see: (a) M. Tadesse, M. B. Strom, J. Svenson, M. Jaspars, B. F. Milne, V. Torfoss, J. H. Andersen, E. Hansen, K. Stensvag and T. Haug, Org. Lett., 2010, 12, 4752–4755 CrossRef CAS PubMed; (b) P. García, M. Martín-Pastor, A. R. de Lera and R. Alvarez, Magn. Reson. Chem., 2010, 48, 543–549 CrossRef PubMed; (c) R. H. Contreras, P. F. Provasi, F. P. dos Santos and C. F. Tormena, Magn. Reson. Chem., 2009, 47, 113–120 CrossRef CAS PubMed.
- A. Bax and R. Freeman, J. Am. Chem. Soc., 1982, 104, 1099–1100 CrossRef CAS.
- J. B. Harborne, The Flavonoids, Chapman and Hall, 1988 Search PubMed.
- (a) G. Blaskó, L. Xun and G. A. Cordell, J. Nat. Prod., 1988, 51, 60–65 CrossRef; (b) J. Budzianowski, M. Morozowska and M. Wesołowska, Phytochemistry, 2005, 66, 1033–1039 CrossRef CAS PubMed; (c) G. P. Romanelli, E. G. Virla, P. R. Duchowicz, A. L. Gaddi, D. M. Ruiz, D. O. Bennardi, E. del Valle Ortiz and J. C. Autino, J. Agric. Food Chem., 2010, 58, 6290–6295 CrossRef CAS PubMed.
- (a) S. Venkateswarlu, G. K. Panchagnula, A. L. Gottumukkala and G. V. Subbaraju, Tetrahedron, 2007, 63, 6909–6914 CrossRef CAS; (b) Y. Usami, Mar. Drugs, 2009, 7, 314–330 CrossRef CAS PubMed.
- J. L. Marshall, Carbon–Carbon and Carbon–Proton NMR Couplings: Applications to Organic Stereochemistry and Conformational Analysis, Verlag Chemie International, Deerfield Beach, FL, 1983, pp. 40–41 Search PubMed.
- B. A. Keay, J. M. Hopkins and P. W. Dibble, Furans and their Benzo Derivatives: Applications. Comprehensive Heterocyclic Chemistry III, Elsevier, The Netherlands, 2008 Search PubMed.
- (a) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084–3213 CrossRef CAS PubMed; (b) W. J. Moran and A. Rodríguez, Org. Prep. Proced. Int., 2012, 44, 103–130 CrossRef CAS.
- M. Hoffmann, S. Miaskiewicz, J.-M. Weibel, P. Pale and A. Blanc, Beilstein J. Org. Chem., 2013, 9, 1774–1780 CrossRef PubMed.
- N. Shanker, O. Dilek, K. Mukherjee, D. W. McGee and S. L. Bane, J. Fluoresc., 2011, 21, 2173–2184 CrossRef CAS PubMed.
- R. Kazlauskas, P. T. Murphy, R. J. Quinn and R. J. Wells, Tetrahedron Lett., 1977, 18, 37–40 CrossRef.
- A. J. Frontier and C. Collison, Tetrahedron, 2005, 61, 7577–7606 CrossRef CAS.
- (a) M. Hoffmann, J.-M. Weibel, P. de Frémont, P. Pale and A. Blanc, Org. Lett., 2014, 16, 908–911 CrossRef CAS PubMed; (b) M.-C. Cordonnier, A. Blanc and P. Pale, Org. Lett., 2008, 10, 1569–1572 CrossRef CAS PubMed.
- (a) W. He, X. Sun and A. J. Frontier, J. Am. Chem. Soc., 2003, 125, 14278–14279 CrossRef CAS PubMed; (b) M. Janka, W. He, A. J. Frontier, C. Flaschenriem and R. Eisenberg, Tetrahedron, 2005, 61, 6193–6206 CrossRef CAS; (c) M. Janka, W. He, I. E. Haedicke, F. R. Fronczek, A. J. Frontier and R. Eisenberg, J. Am. Chem. Soc., 2006, 128, 5312–5313 CrossRef CAS PubMed; (d) J. Huang and A. J. Frontier, J. Am. Chem. Soc., 2007, 129, 8060–8061 CrossRef CAS PubMed; (e) W. He, I. R. Herrick, T. A. Atesin, P. A. Caruana, C. A. Kellenberger and A. J. Frontier, J. Am. Chem. Soc., 2007, 130, 1003–1011 CrossRef PubMed.
- The factors causing this effect are not clear but were already observed and discussed: see: G. J. Karabatsos, J. Am. Chem. Soc., 1961, 83, 1230–1232 CrossRef CAS.
- A. Vij, Y. Y. Zheng, R. L. Kirchmeier and J. M. Shreeve, Inorg. Chem., 1994, 33, 3281–3288 CrossRef CAS.
- A. K. Al-Sa'Ady, C. A. Mcauliffe, R. V. Parish, J. A. Sandbank, R. A. Potts and W. F. Schneider, in Inorganic Syntheses, ed. S. Kirschner, John Wiley & Sons, Inc., 1985, pp. 191–194 Search PubMed.
- N. Mézailles, L. Ricard and F. Gagosz, Org. Lett., 2005, 7, 4133–4136 CrossRef PubMed.
- N. P. Cheval, A. Dikova, A. Blanc, J.-M. Weibel and P. Pale, Chem.–Eur. J., 2013, 19, 8765–8768 CrossRef CAS PubMed.
- M. Yoshida, Y. Fujino and T. Doi, Org. Lett., 2011, 13, 4526–4529 CrossRef CAS PubMed.
- M. Yoshida, Y. Fujino, K. Saito and T. Doi, Tetrahedron, 2011, 67, 9993–9997 CrossRef CAS.
- A. W. Sromek, A. V. Kel'in and V. Gevorgyan, Angew. Chem., Int. Ed., 2004, 43, 2280–2282 CrossRef CAS PubMed.
- T. Schwier, A. W. Sromek, D. M. L. Yap, D. Chernyak and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 9868–9878 CrossRef CAS PubMed.
- A. Arcadi, A. Burini, S. Cacchi, M. Delmastro, F. Marinelli and B. R. Pietroni, J. Org. Chem., 1992, 57, 976–982 CrossRef CAS.
- K. Ji, J. Nelson and L. Zhang, Beilstein J. Org. Chem., 2013, 9, 1925–1930 CrossRef PubMed.
- (a) G. Liang, S. N. Gradl and D. Trauner, Org. Lett., 2003, 5, 4931–4934 CrossRef CAS PubMed; (b) M. Rueping, W. Ieawsuwan, A. P. Antonchick and B. J. Nachtsheim, Angew. Chem., Int. Ed., 2007, 46, 2097–2100 CrossRef CAS PubMed.
- C. Bee, E. Leclerc and M. A. Tius, Org. Lett., 2003, 5, 4927–4930 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional schemes, crystallographic structure refinements of compound 10a and spectral data. CCDC 1047510. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra03228h |
| This journal is © The Royal Society of Chemistry 2015 |