
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Review of the bulk and surface chemistry of iron in atmospherically relevant systems containing humic-like substances
Hind A. Al-Abadleh
*
Department of Chemistry and Biochemistry, Wilfrid Laurier University, Waterloo, ON N2L 3C5, Canada. E-mail: halabadleh@wlu.ca; Tel: +1-519-884-0710 ext. 2873
First published on 6th May 2015
Abstract
As the fourth most abundant element by mass in the Earth's crust, iron is ubiquitous and its chemistry is rich and interdisciplinary in nature. This review synthesizes the current state of knowledge of iron chemistry in multicomponent atmospheric aerosols. This knowledge is also applicable to other atmospherically relevant systems that include iron-containing anthropogenic nanodust, ocean surfaces and buildings. Because of the abundance of humic-like substances in these systems, this review focuses on the chemistry of these substances with iron compounds. Findings from field measurements and laboratory studies are summarized to highlight the major themes in the chemical reactivity of iron, which varies depending on the solubility, the redox conditions, the absence and presence of UV-visible light and reactive oxygen species, the pH and the temperature. This review also highlights the key differences between the bulk and surface chemistry of iron-containing materials, which varies considerably because of the structure of the interfacial water and the solvent cage effect. Additional laboratory, field and modelling studies are needed to better understand the contributions of transition metal chemistry to the formation of secondary organic aerosols and also the chemistry, uptake and release of trace gas phase species. This information will improve the predictive power of models that incorporate aerosol chemistry and physics.
 Hind A. Al-Abadleh | Hind A. Al-Abadleh is currently an Associate Professor of physical chemistry at Wilfrid Laurier University. Her research interests include heterogeneous photochemistry in atmospheric aerosols driven by transition metals and the surface chemistry of arsenic and phosphorus compounds in model soil systems. She has received an Early Researcher Award from the Ontario Ministry of Research and Innovation and the Petro-Canada Young Innovator Award, among other awards. Al-Abadleh completed her BSc (Hons) degree in chemistry at the United Arab Emirates University (1999), followed by a PhD at the University of Iowa with Professor Vicki Grassian (2003) and postdoctoral training with Professor Franz Geiger at Northwestern University (2005). |
1. Introduction
Atmospheric aerosols impact the Earth's climate directly through their ability to absorb and scatter solar radiation and also indirectly by affecting the formation and lifetime of clouds and by influencing the concentration and type of gas phase species by providing surfaces for them to react on.1 The deposition of aerosols also affects the Earth's biota and the biogeochemistry of certain metals.2–5 The Fifth Assessment Report of the Intergovernmental Panel on Climate Change showed that the representation of aerosols in climate models is still inadequate as a result of their highly complex physicochemical properties that change over time.6 This large uncertainty associated with aerosols is a result of the relatively low level of scientific understanding of their indirect effects on climate, i.e. their role in acting as condensation nuclei for clouds and ice.7 Specifically, climate and cloud chemistry models need experimental data on the hygroscopic properties of aerosols,8,9 secondary pathways for their formation10,11 and the rates of heterogeneous photochemical reactions.12–14 The uptake of water by aerosols is controlled by their chemical composition and interfacial properties and is sensitive to changes in meteorological parameters such as the relative humidity (RH).8,15 The heterogeneous photochemical reactivity of aerosols causes the degradation of their organic content, the formation of secondary organic aerosols (SOAs) and also enhances the uptake and release of gas phase species.13,14,16–19One important class of organic matter in atmospheric aerosols is humic-like substances (HULIS).20,21 As a result of their complex chemical nature, HULIS in aerosols have been studied less than the other classes of organic compounds identified in atmospheric aerosols. As noted by Zetzsch and co-workers,22,23 the lack of information about the true composition of HULIS is underestimated. Atmospheric HULIS are emitted from primary sources such as wind-blown marine sediments, soils and the burning of biomass, or are formed in the atmosphere through condensation and polymerization.20,21,24 They comprise 15–60% of the water-soluble organic carbon (WSOC) in aerosols. Duarte et al.24 found from infrared (IR) and solid-state 13C-NMR spectroscopy that atmospheric HULIS contain conjugated carbonyl groups, ethers, aromatic phenols, carboxyl groups, alcohols, oxygenated aliphatic carbon, branched alkyl chains and a high level of aromatic and aliphatic content. As a result, most laboratory studies so far have used surrogates for atmospheric HULIS, such as humic and fulvic acids from terrestrial and aquatic sources,25,26 tannic acid (1,2,3,4,6-pentagalloyl-O-glucose, C76H52O46),26–28 gallic acid (3,4,5-trihydroxybenzoic acid),29 catechol (1,2-dihydroxybenzene),23,30,31 guaiacol (2-methoxyphenol)23,32 and shikimic acid.33 The latter three compounds are semi-volatile phenolic compounds emitted from the burning of biomass and are known aromatic SOA precursors.23,33
The hygroscopic growth of these model compounds has been studied using a number of techniques, including electrodynamic balance and hygroscopic tandem differential mobility analysis in both their pure form and when mixed with inorganic salts,34,35 and insoluble mineral aerosol,35 IR spectroscopy36 and X-ray techniques.26 These studies found that surrogates for HULIS undergo growth and evaporation as a function of the RH, which changes their size. These model systems are also efficient cloud condensation nuclei, can lower or enhance water uptake when mixed with soluble salts34 and can enhance the adsorption of water when coating insoluble calcite particles.35 Experiments on the hygroscopic properties of HULIS in aerosols showed continuous water uptake as a function of the RH, with the exception of fulvic acid, which showed phase separation.26,34,37,38 The structure of water on some of these systems resembled that of water at the interface with polar organic solvents.36 The amount of water uptake was found to vary depending on the carbon functional groups in the fulvic acid samples after phase separation.26 This means that, depending on the size and water content of the particles, preferential partitioning of HULIS to the surface could become important in their overall surface reactivity. Although bulk photochemical reactions involving humic substances in atmospheric aerosols and aquatic systems have received much attention,13,14,39–41 fewer studies have reported the heterogeneous photochemistry of atmospheric HULIS. Examples include: (a) the fast photosensitized formation of HONO from the photoreaction of NO2 with humic,42,43 tannic and gentisic acid films28 under dry and humid conditions; (b) the photooxidation, formation and characterization of secondary particles from catechol and guaiacol;23 and (c) changes to the functional groups characteristic of solid tannic acid and the formation of new carbonyl groups characteristic of aryl aldehydes and/or quinone under humid conditions.36
The chemical transformation and aging of aerosols due to interaction with atmospheric oxidants and other trace gases proceed differently depending on their phase state and diffusivity.44–46 The term “chemical aging of aerosols” is used to describe the transformation of organic matter in atmospheric particles that occurs via: (a) heterogeneous reactions with reactive radicals such as O3, ˙OH and ˙NO3 and acidic gases such as HNO3 and HSO4; (b) the condensation of semi-volatile organic compounds such as phenols and organic nitrates; (c) particle phase reactions resulting in the formation of oligomers, such as aldol addition reactions; and (d) heterogeneous photosensitized reactions.47–51 This latter pathway could be driven by the electronically excited states of some organic chromophores such as HULIS or by soluble and insoluble transition metals such as iron.14
In addition to influencing the oxidative power of the atmosphere and the global sulfate budget,41,52 particulate iron can catalyse reactions that lead to oxidative stress in living cells as a result of the production of reactive oxygen species (ROS).53 Dark and photochemical processes that lead to the release of soluble iron into the aqueous phase have consequences for the availability of this essential element for phytoplankton productivity, the extent of algal blooms and the uptake of atmospheric CO2.54,55
The objective of this review is to synthesize the current state of knowledge on the role of iron in the chemical aging of atmospheric aerosols containing organic matter, particularly those that model HULIS. The review is organized into five main sections and starts with a synthesis of recent results from field measurements and modelling studies, followed by a summary of the highlights of iron chemistry under dark conditions and the photochemical reactions driven by iron obtained from bulk and surface-sensitive measurements. Because of the molecular level differences between bulk and surface water, a concise summary of the literature on the latter topic is also provided. The review concludes with a summary and directions for future research.
2. Field measurements and modelling studies of iron in aerosols
2.1. Sources and chemical characterization
The source of the iron added to ocean surfaces is mainly mineral dust,2,56,57 with contributions from anthropogenic emissions58 and the burning of biomass.59,60 After the deposition of dust, iron undergoes dissolution and complexation with organic matter. These processes are affected by exposure to light and hence affect the fate and cycling of this element.61–63 Other sources of the dissolved iron added to the oceans include hydrothermal vents and reductive and non-reductive release from oceanic sediments. Conway and John57 analysed samples along a section of the North Atlantic Ocean to determine the source-sensitive dissolved stable iron isotope ratios (δ56Fe) and the iron concentration [Fe]. The results showed that 71–87% of dissolved iron in the North Atlantic Ocean originated from Saharan dust aerosols, 10–19% from the non-reductive release of iron from oxygenated sediments, 1–4% from the reductive dissolution of sediments and 2–6% from hydrothermal venting.Iron is a limiting nutrient for phytoplankton in about 40% of the ocean.64,65 The estimated annual amount of total dissolved iron deposited on ocean surfaces from mineral dust is 24 × 109 mol per year,66 which is unevenly distributed and tends to be concentrated in the tropical Atlantic Ocean, the Indian Ocean, the Mediterranean Sea and the Arctic.67 In a related study, von der Heyden et al.68 analysed the size, iron oxidation state, and the composition and degree of crystallinity of iron-containing particles in the waters of the Southern Ocean euphotic zone from South Africa to Antarctica. High-resolution images and synchrotron-based X-ray spectra showed that the particles ranged in size from 20 to 700 nm, with a variety of phases that included Fe(III) and Fe(II) (oxyhydr)oxides, in addition to mixed phases of both elements. The relative concentration of each iron-rich particle phase varied depending on the sample location. For example, the samples closest to the African continent showed the largest degree of chemical heterogeneity and consisted mostly of Fe(III)-rich particles. This was in contrast with the samples from south of the southern boundary, where the samples were mostly rich in Fe(II). The ratio Al![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe was also found to vary with the depth and the distance from land sources to the open ocean. References to aluminium are made as this element is considered to be a solubility modifier of Fe(III) minerals and an indicator of the source of particles. High Al:Fe ratios (0–0.47) are usually found in weathered minerals inland as a result of the substitution of aluminium with iron and are less soluble than the unsubstituted minerals. In this study,68 higher Al:Fe values were found in deeper waters than in surface waters (up to 0.17) and nearer to the land (up to 0.2); these ratios decreased towards the open ocean. The study also compared the average summer chlorophyll a concentration with the abundance and distribution of labile forms of iron, reflecting the effect of iron speciation on biological systems.
:Fe was also found to vary with the depth and the distance from land sources to the open ocean. References to aluminium are made as this element is considered to be a solubility modifier of Fe(III) minerals and an indicator of the source of particles. High Al:Fe ratios (0–0.47) are usually found in weathered minerals inland as a result of the substitution of aluminium with iron and are less soluble than the unsubstituted minerals. In this study,68 higher Al:Fe values were found in deeper waters than in surface waters (up to 0.17) and nearer to the land (up to 0.2); these ratios decreased towards the open ocean. The study also compared the average summer chlorophyll a concentration with the abundance and distribution of labile forms of iron, reflecting the effect of iron speciation on biological systems.
A number of studies have focused on single-particle analysis of field-collected aerosols from marine, urban and rural sites, particularly those containing soluble and insoluble iron.69–80 Schroth et al.63 demonstrated that iron speciation (the oxidation state and bonding environment) varies with the source of the aerosol. For example, soils in arid regions are dominated by Fe(III) (oxyhydr)oxides, glacially weathered particles by Fe(II) silicates, and oil fly ash from fossil fuel combustion by Fe(III) sulfates. As a result, variations in iron solubility were observed among these aerosol types, which increased in this order: arid soils (<1% of iron was soluble), glacial products (2–3% iron soluble) and oil fly ash (77–81% of iron soluble). Fig. 1 shows the iron signature associated with biological material in a representative positive ion mass spectrum of individual sea spray particles analysed by aerosol time-of-flight mass spectrometry (ATOFMS).76 When the pH of the droplet was acidic, the organic material was concentrated on the surface as an outer layer.81 The chemical and physical properties of marine primary organic aerosols and their impact on the Earth's climate have been reviewed elsewhere.82 The transport of transition metals such as iron from the oceans to the atmosphere and their enrichment at the interface of marine aerosols have also been known for some time.83–85
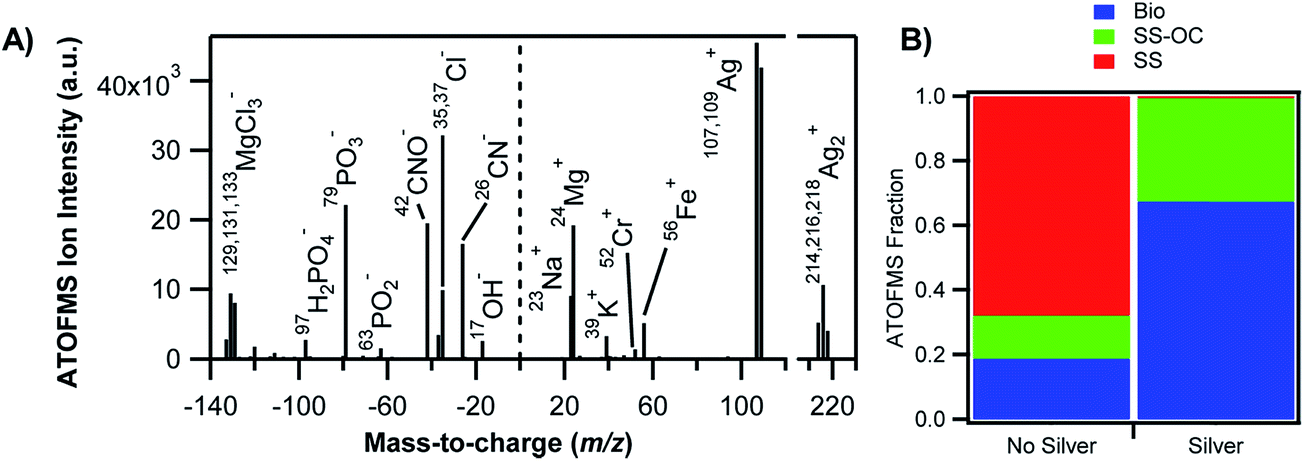 | ||
| Fig. 1 Association of transition metals with biological material in sea spray aerosols. (A) Negative (left) and positive (right) mass spectra of a representative individual silver-containing particle. (B) Relative contributions of the different particle types as measured by aerosol time-of-flight mass spectrometry (ATOFMS) for particles without silver (left) and silver-containing particles (right). Bio = bioparticles, SS-OC = sea salt with organic carbon, SS = sea salt. Reprinted with permission from ref. 76 (Copyright © 2014, American Chemical Society). | ||
The speciation of iron has been determined from single-particle analysis in a number of urban and rural sites in the USA, including Michigan,74 Georgia,69 Ohio75 and Los Angeles.77 Using a particle-into-liquid sample coupled with a liquid waveguide capillary cell and UV-visible spectrophotometry, Oakes et al.74 measured fine particle (PM2.5) water-soluble ferrous iron [WS_Fe(II)] in Dearborn, Michigan and Atlanta, Georgia. The concentrations ranged from 4.6 to 400 ng m−3, the highest concentrations of which were found to be associated with sulfate plumes and those with highest apparent aerosol acidity. The temporal trends in WS_Fe(II) were found to be linked with industrial emissions or atmospheric processing of these emissions leading to the formation of WS_Fe(II). In another study by the same group,69 X-ray absorption near-edge structure spectroscopy and microscopic X-ray fluorescence were used to measure the mineralogy and oxidation state of iron in single particles collected from urban and rural sites in Georgia. These measurements were complemented by experiments to determine the fractional solubility of iron. The X-ray absorption near-edge structure spectroscopy measurements showed that iron was present as a mixture of Fe(II) and Fe(III), with the majority of the particles (74%) characterized as Al-substituted Fe oxides followed by Fe aluminosilicates (12%). The spatial distribution of iron in coarse-mode particles was studied using X-ray elemental mapping and computer-controlled scanning electron microscopy.75 Fig. 2 shows the scanning electron microscopy images and energy-dispersive X-ray analysis elemental maps of representative iron-containing particles from Cleveland, Ohio, which were classified as fly ash, mineral dust, NaCl-containing and Ca–S-containing agglomerates (top to bottom). The concentration of iron-containing particles peaked in the 3–6 μm diameter range to about 5 μg m−3. Fly ash particles are characteristic of industrial emissions (e.g. steel production)86 and their concentration was found to decrease strongly with the distance from the source emissions.75 As noted by the authors,75 the Cleveland study highlights the importance of accounting for anthropogenic sources of iron-containing particles to explain soluble iron (FeS) concentrations that exceed estimates based solely on mineral dust aerosols.87
 | ||
| Fig. 2 Scanning electron microscopy images and energy-dispersive X-ray analysis elemental maps of particles representing (a) fly ash, (b) mineral dust, (c) NaCl agglomerates and (d) Ca–S agglomerates. Reprinted with permission from ref. 75 (Copyright © 2012, American Chemical Society). | ||
Using single-particle aerosol time-of-flight mass spectrometry, the dynamic size and chemical composition of aerosols were measured near Los Angeles and in the Long Beach Port region.77 Particles in the 0.1–1 μm size range showed the characteristic signatures of soot, i.e. the transition metals iron, vanadium and nickel associated with sulfate and nitrate. The sources of these particles were attributed to primary emissions from oil combustion in ships, refineries and traffic in the port region, in addition to secondary processing during transport. The particle concentration ranged from 14 to 44 μg m−3 over the collection time. As stated by the authors,77 the significance of this study lies in the fact it highlighted that the aforementioned primary sources need to be regulated to improve the air quality in California, as for car and truck emissions.
Deboudt et al.79 analysed thousands of individual particles collected during a campaign off the Atlantic coast of West Africa during the African monsoon, where the sampling sites were located on the path of the Saharan dust plumes. The study aimed to analyse the particle size, morphology, chemical composition and mixing states using scanning electron microscopy-energy-dispersive X-ray spectrometry, transmission electron microscopy-electron energy loss spectroscopy and Raman microspectrometry. The three classes of aerosols – mineral dust, carbonaceous compounds and marine compounds – were predominantly externally mixed. About 10.5–46.5% of the analysed particles were found to be internally mixed binary systems, i.e. dust/carbonaceous, carbonaceous/marine and dust/marine. The mineral content included iron oxides, aluminosilicates and Mg aluminosilicates, calcium carbonate, gypsum, titanium oxide and silica. The carbonaceous content originated from the burning of biomass (tar balls) and anthropogenic (soot) emissions and was found to be amorphous in structure rather than crystallized carbon. The results also revealed that marine and carbonaceous compounds generally formed a coating of mineral dust particles.
Moreover, because the oxidation state and morphology of iron-containing aerosols can modify the chemical reactivity, Takahama et al.80 performed a comprehensive analysis of 63 iron-containing particles from five field campaigns using near-edge X-ray absorption fine structure L-edge spectroscopy coupled with scanning transmission X-ray microscopy. Data generated from this method detected heterogeneities in the distribution of iron and the redox state over individual particles. These particles ranged in size from 0.2 to 4.5 μm with ratios of Fe(II) to total iron (FeT) from 0 to 0.73. The X-ray images showed many different morphologies; in some cases there was a clear barrier between the iron and the carbonaceous regions. The variability in the Fe(II) fraction was analysed as a function of the distance from the surface. The results showed higher Fe(II) fractions near the surface than towards the interior, which was consistent with the surface reduction mechanisms for iron.
The complexation of iron and other transition metals such as copper and manganese to organic ligands in size-segregated ambient aerosol particles was investigated by Scheinhardt et al.88 Samples were collected from nine sites in Germany that covered urban, rural and coastal areas. Coupled with aerosol thermodynamic modelling results, the analysis showed that pH, [Ca2+], [Mg2+] and the formation of insoluble Ca oxalate governed the availability of oxalate as the main strong organic ligand that preferentially bound Fe(III). Other factors, such as the season, the origin of the air mass, the sampling site and the particle size, were also found to affect the complexation of transition metals to organic ligands. In summary, the relatively high loading of atmospheric iron-containing dust and the processing pathways with and without organic ligands demand adequate representation in atmospheric models; this representation starts with the mineralogy of the particles.
2.2. Modelling the cycling of iron
Nickovic et al.89 developed a high-resolution dataset (GMINER30) of the mineral composition of potentially dust-producing soils that occupy the majority of the arid regions on a global scale. Fig. 3 shows the global distribution of the effective percentage of minerals in soils around the world. The processes that lead to the generation of atmospheric dust particles by wind action from soils have been reviewed elsewhere and include suspension, creep, saltation and sandblasting.90 In another study by Nickovic et al.,91 a regional atmospheric dust-iron model (DREAM) was developed to numerically simulate the atmospheric route of iron from deserts to sinks in the ocean. This model included the parameterization of the transformation of iron to a soluble form caused by the mineralogy of the dust, cloud processes and solar radiation. The modelling results were compared with observations collected from several Atlantic Ocean cruise routes dominated by dust aerosols. This model underestimated FeT and FeS for reasons that included small iron emissions at the source origins and neglecting the influence of anthropogenic sources and the burning of biomass.91 Using the aerosol chemistry version of Integrated Massively Parallel Atmospheric Chemical Transport (IMPACT), which includes three classes of iron-containing minerals, Ito and Xu92 investigated the deposition of filterable (i.e. soluble) FeS and its response to changes in the anthropogenic emissions of combustion aerosols and precursor gases. They found that the scavenging efficiency of dust particles depended on the surface coating of these aerosols by sulfate, nitrate and ammonium species and that the release of iron from minerals was a function of the acidity of the aerosol. Although this release process could happen readily under highly acidic conditions (pH < 2), the results showed that iron released slowly at higher pH values during long-range transport appeared to be important. This model also projected a decrease in the deposition of filterable iron to the Western North Pacific from iron-containing mineral dust as a result of less acidification in dust from Asia as a result of air quality improvements, which have reduced the emission of nitrogen oxides (NOx). In another modelling study, Ito60 aimed to estimate the atmospheric sources of bioavailable iron; an explicit scheme for the dissolution of iron in combustion aerosols as a result of photochemical reactions with inorganic and organic acids in solution was implemented in an atmospheric chemistry transport model. The results showed that 40% of FeT over major portions of the open ocean in the Southern Hemisphere originated from the deposition of FeS from combustion sources.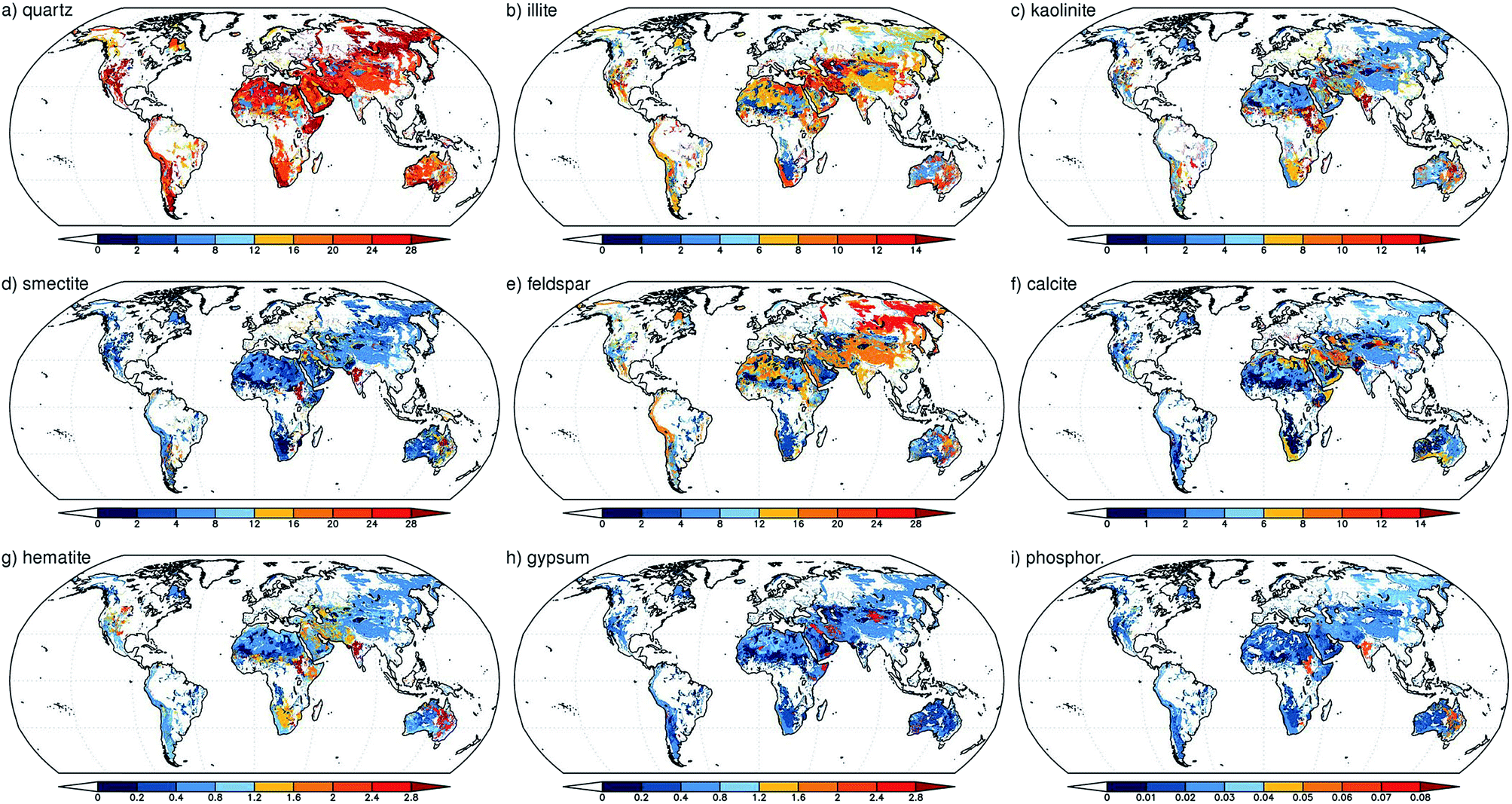 | ||
| Fig. 3 Global distribution of the effective mineral content of soils (as a percentage) for: (a) quartz; (b) illite; (c) kaolinite; (d) smectite; (e) feldspar; (f) calcite; (g) hematite; (h) gypsum; and (i) phosphorus. The mineral fraction was weighted with the clay and silt content of the soil. For minerals that were present in both clays and silts, the weighted values were summed. Reproduced with permission from ref. 89. | ||
Sholkovitz et al.93 provided a synthesis of a global- and regional-scale dataset on the fractional solubility of aerosol iron (%FeS) relative to FeT from over 1000 samples collected from a number of sites, including open and coastal ocean sites and some continental sites. The trend that emerged was described by a simple two-component mixing model, where %FeS reflected the mixing of mineral dust (high FeT and low %FeS) and aerosols from combustion sources (low FeT and high %FeS).93 More sophisticated global chemical transport models were used for the same purpose, such as the GEOS-Chem94 and IMPACT95 models. For example, the former model was used to analyse the magnitude and spatial distribution of mineral dust and FeS deposition to the South Atlantic Ocean.94 The model predicted a <1% dissolved iron fraction of mineral dust over the South Atlantic Ocean as a result of the low ambient concentrations of acidic trace gases available for mixing with dust plumes. Sensitivity studies showed that the initial amount of FeS in the dust source regions to a large extent controls the amount of FeS deposited to the South Atlantic Ocean.94 In another study, the effect of aerosol emissions from ship plumes on the solubility of iron in particles from combustion sources was modelled over the high-latitude North Atlantic and North Pacific Oceans using IMPACT.95 The results showed that combustion particles with a low iron loading (1–110 ng m−3) contributed more than 10% of the FeS. This model predicted that, in the year 2100, FeS from ships could contribute 30–60% of the total FeS deposition. Before deposition and while suspended in the atmosphere, Guo et al.96 found from field measurements in Hong Kong and modelling results that aqueous phase reactions involving transition metal ions such as iron, copper and manganese in deliquescent aerosols significantly affected the mixing ratios of H2O2 in the gas phase by acting as net sources or sinks depending on the metal content of the aerosols.
In summary, these field studies were aimed at collecting, analysing and tracking the fate of iron in atmospheric aerosols from different sources. The studies summarized here clearly show the impact that the mineralogy and the atmospheric and surface ocean processing of field-collected aerosols have on the availability of soluble iron and its association with organic matter. Such results provide a framework for improving the representation of iron-containing particles in models. They also highlight the importance of understanding the chemical reactivity of iron at the molecular level under various conditions, which is the subject of the following sections.
3. Bulk chemistry of soluble iron in the dark
3.1. Speciation of bulk iron in the dark
Iron chemistry in the dark provides a potentially important abiotic pathway for the night-time oxidation of organic compounds that could compete with nitrate radical chemistry.97 In the bulk aqueous phase, the chemistry of iron depends on which species is present at a given pH. The bulk speciation curves of Fe(III) in solution show that the pH affects the concentration of the hydrated species [Fe(H2O)6]3+, [Fe(H2O)5(OH)]2+ and [Fe(H2O)4(OH)2]+.98–100 In the presence of halides, such as chloride (Cl−) ions, species such as [Fe(H2O)5Cl]2+ and [Fe(H2O)4Cl2]+ also exist (Fig. 4). Curves shown in Fig. 4 are generated by the following reactions and equilibrium constants at pH ≤ 3 and 25 °C:99| [Fe(H2O)6]3+ ⇌ [Fe(H2O)5(OH)]2+ + H+, K1 = 6.5 × 10−3 M | (1) |
| [Fe(H2O)6]3+ ⇌ [Fe(H2O)4(OH)2]+ + 2H+, K2 = 2.1 × 10−6 M2 | (2) |
| [Fe(H2O)6]3+ + Cl− ⇌ [Fe(H2O)5Cl]2+ + H2O, K3 = 30.2 M−1 | (3) |
| [Fe(H2O)6]3+ + Cl− ⇌ [Fe(H2O)4(Cl)2]+ + 2H2O, K4 = 134.9 M−2 | (4) |
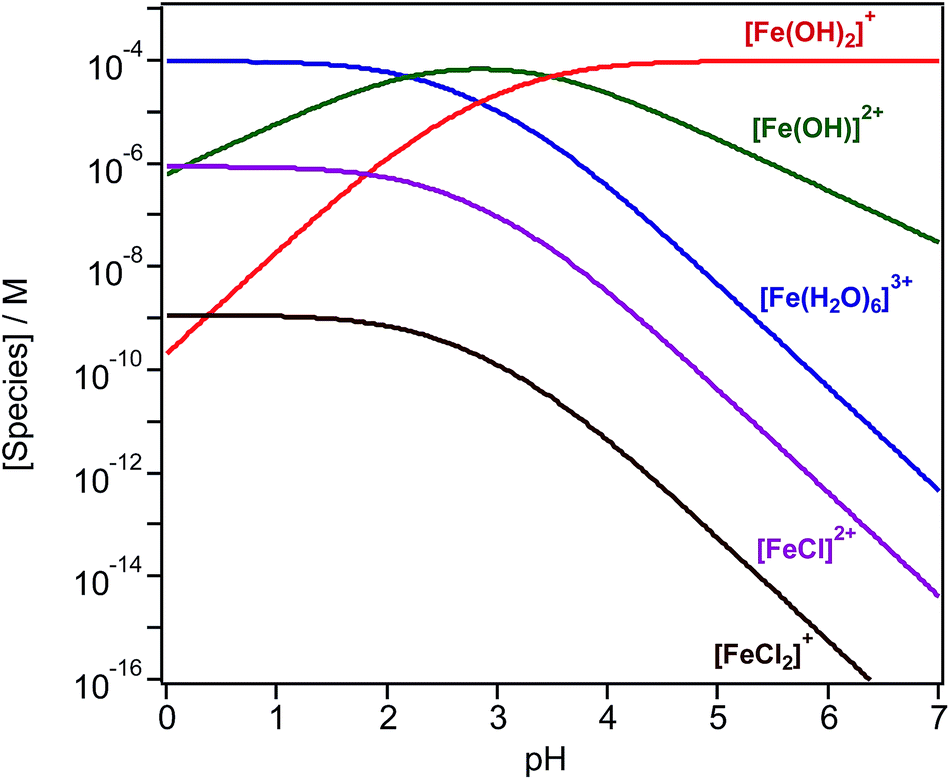 | ||
| Fig. 4 Speciation curves generated for 1 mM FeCl3 solution as a function of pH at 25 °C, plotted on a log scale, using an in-house MATLAB program courtesy of Dr D. Scott Smith, Wilfrid Laurier University. | ||
In addition to Cl− ions, the presence of other inorganic and organic species affects the speciation of Fe(III). Wittmer et al.101 have listed equilibrium constants for reactions with bromide, sulfate, oxalate and catechol, which could be used in constructing curves such as those in Fig. 4. The following sections highlight a number of examples of chemical reactions driven by these iron species.
3.2. Generation of reactive oxygen species and further reaction with organic compounds
Fenton reactions, driven mainly by the addition of H2O2 to bulk aqueous solutions containing Fe(II), are efficient in degrading soluble organic matter as a result of the formation of hydroxyl radicals, as shown in reaction (5):102–107| Fe(II) + H2O2 → Fe(III) + ˙OH + OH−, k = 55 M−1 s−1 | (5) |
In addition, these reactions can form other ROS such as the superoxide anion (O2˙−), which explain the harmful effects to human health of inhaling metal-containing aerosols.53,108–114 As shown in the following photochemistry sections, light accelerates the recycling of Fe(III) to Fe(II) in a system containing H2O2 according to reaction (6):107
| Fe(III) + H2O2 → Fe(II) + HO2˙/O2˙− + H+, k = 2.00 × 10−3 M−1 s−1 | (6) |
Organic species such as quinones and carboxylate ligands were shown to have a similar effect.107,115 Even in chemically large compounds such as HULIS, the high density of the carboxylate and quinoid units have been shown to enhance the degradation of pyrene and phenol – used as model organic pollutants released by anthropogenic sources – by promoting dark Fenton reaction (5) in the aqueous phase.116
A number of studies have identified the degradation products of gallic acid and catechol by Fenton's reagent, H2O2 and Fe(II). Duesterberg and Waite107 reported a kinetic model based on experimental data that included reaction rate constants for the degradation of gallic acid by Fe(III). On complexation with Fe(III), a gallic acid-semiquinone species formed, which, in the presence of Fe(III)/Fe(II) resulted in the formation of a gallic acid-quinone compound. An attack by ˙OH radicals on the latter species resulted in ring opening and then further reactions until mineralization was complete. In a study by Zazo et al.,104 catechol was formed first from the oxidation of phenol, which then underwent ring opening. Aliphatic C2–C4 organic acids were detected at later stages, such as maleic acid, which was identified as the primary product from ring cleavage, and fumaric acid (C4). Further oxidation to oxalic (C2) and formic (C1) acids was observed when high Fe(II) and H2O2 concentrations were used. Similar results were reported by M'hemdi et al.117 on the role of Fenton and photo-Fenton processes in the complete mineralization of catechol. Using attenuated total internal reflectance Fourier transform infrared (ATR-FTIR) spectroscopy, Arana et al.118 studied the photo-Fenton degradation of phenol, which resulted in the formation of catechol and hydroquinone as intermediates. It was suggested from their results that pyrogallol formed as a result of the further degradation of catechol. These latter two compounds complexed with Fe(III), lowering its free concentration, and, subsequently, the reaction progress. Fenton-like reactions driven by the combination of Fe(III) and H2O2 in the presence of Cl− ions were reported as key ingredients in the formation of short-lived volatile carbon suboxide (C3O2)119 and volatile trichloromethane (CHCl3) from catechol120 over long time frames exceeding 24 h. The formation of oxalic acid in solution was reported as a primary process after 1 h from the dark oxidation of catechol and other hydroxylated benzenes, a process that was also observed in soils rich in organic matter with spiked Fe(III) and H2O2.121 Fig. 5 summarizes these results and shows a suggested degradation pathway based on these findings and other studies.
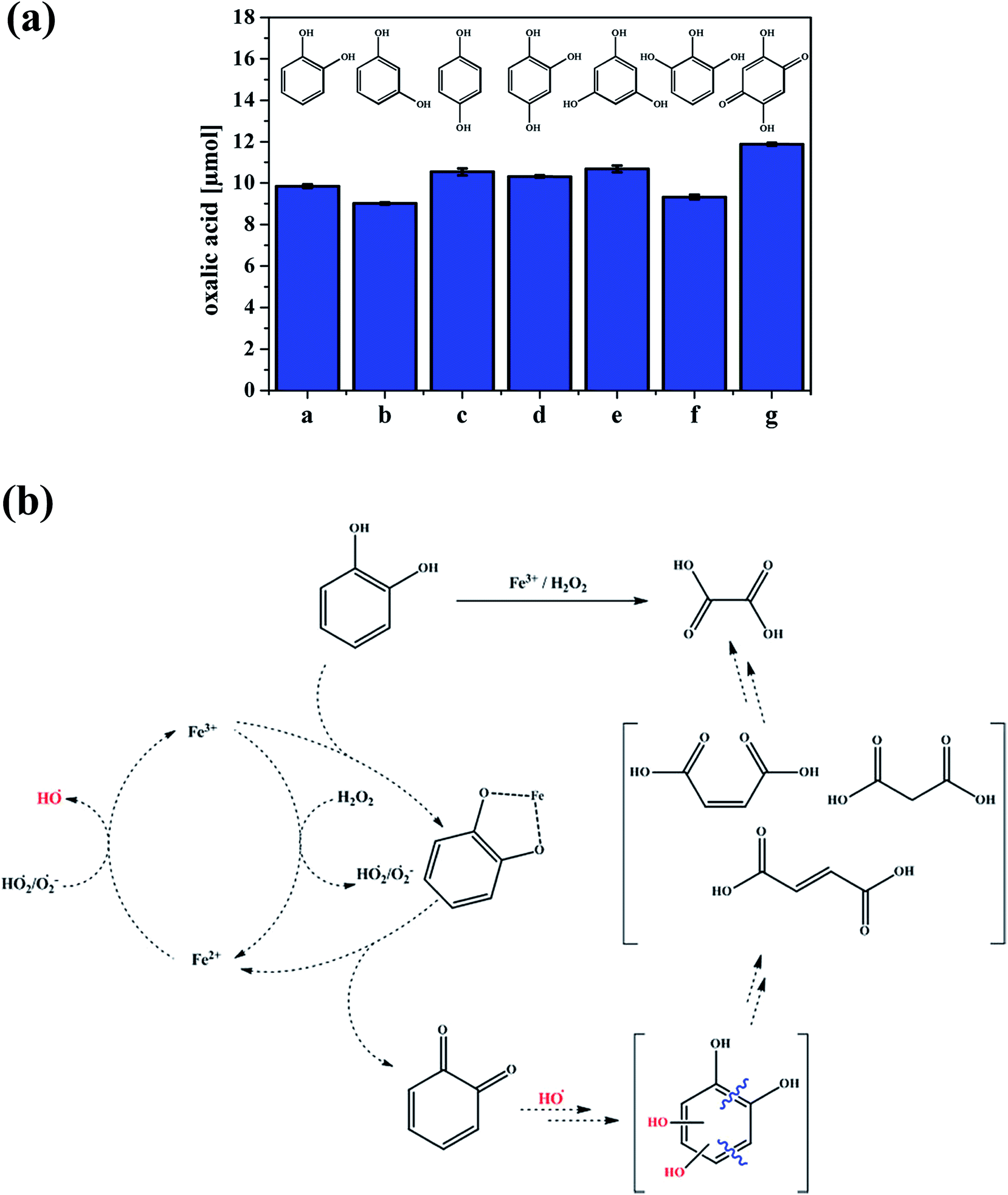 | ||
| Fig. 5 (a) Production of oxalic acid from the reaction of different hydroxylated benzenes (a–g, 0.5 mM) under the conditions of 1.0 mM Fe2(SO4)3 and 7.5 mM H2O2 at 40 °C after 1 h. a = catechol, b = resorcinol, c = hydroquinone, d = 1,2,4-trihydroxybenzene, e = phloroglucin, f = pyrogallol and g = 2,5-dihydroxy-1,4-benzoquinone. (b) Suggested pathway for the production of oxalic acid from catechol. Reproduced with permission from ref. 121 (Copyright © 2013, American Chemical Society). | ||
In addition, Fig. 5b shows that the complexation of Fe with an organic ligand with the catechol moiety results in thermal oxidation in the bulk aqueous phase to form quinone. The spontaneous oxidation of polyphenols to the corresponding quinone, according to reaction (7), proceeds with an oxidation potential of about −0.7 V:122,123
| Polyphenol → o-quinone + nH+ + nē | (7) |
For example, the values for the oxidation of gallic acid, catechol and pyrogallol to the corresponding o-quinone are −0.799, −0.792 and −0.713 V, respectively.122 Despite being a spontaneous reaction for these chemicals, this is a kinetically controlled reaction and is very slow in the presence of dissolved oxygen alone.122 The addition of Fe(III) speeds up this oxidation because the reduction potential to Fe(II) is 0.749 V. The quinone species was found to be relatively unstable in solutions containing gallic acid under acidic conditions.124 Catechol oxidation produces semiquinone and quinone species through a two-electron process (i.e. n = 2 in reaction (7)).123,125 In these studies, the quinone was mainly identified using UV-visible spectrophotometry with its signature broad and relatively weak absorption around 400 nm. This wavelength is very close to that calculated from the HOMO–LUMO gap (3.23 eV) of o-quinone with tert-butyl substituents.126 Fig. 6 shows the UV-visible spectra of gallic acid and catechol on mixing with Fe(III) in the dark under acidic conditions.31 This formation of quinone species is accompanied by the reduction of Fe(III) to Fe(II), which, in oxygenated solution, undergoes autoxidation back to Fe(III) stabilized by complex formation with catechol-containing molecules.122,123
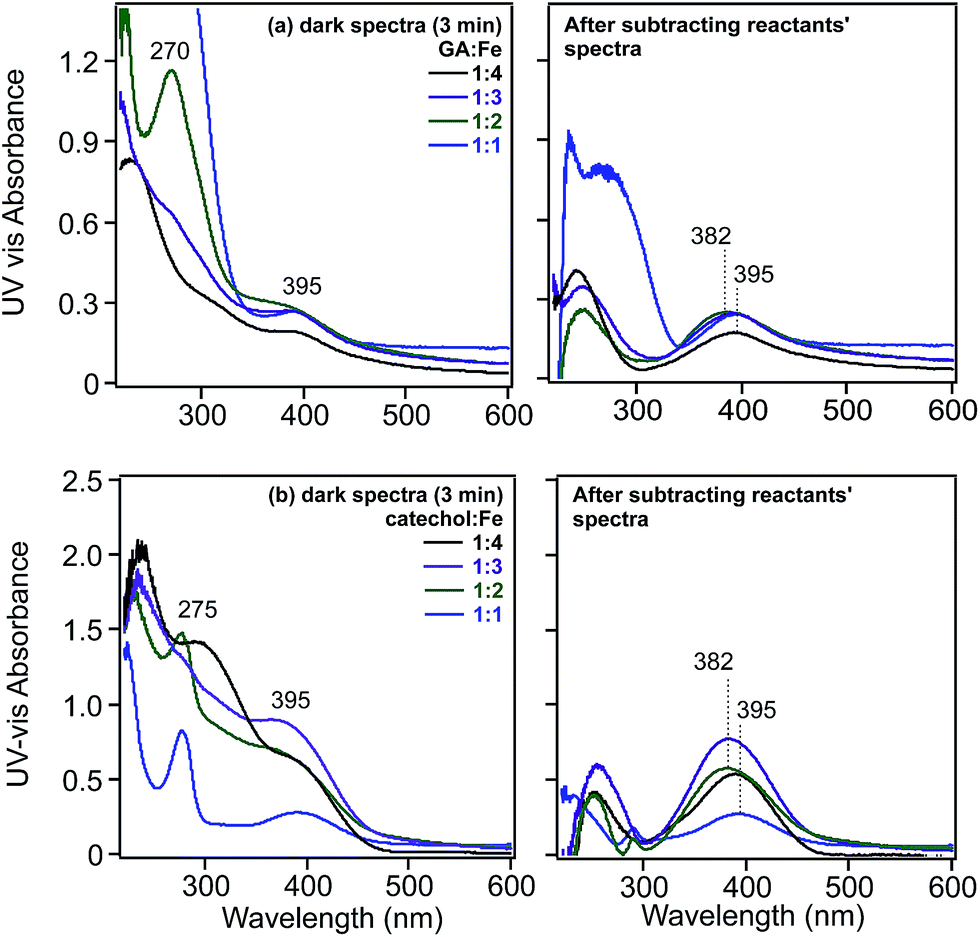 | ||
| Fig. 6 Left-hand panels: UV-visible absorbance spectra of solutions collected after mixing (a) gallic acid and (b) catechol with FeCl3 solutions for 3 min as a function of increasing molar ratios of Fe. Right-hand panels: resultant spectra of reaction mixtures after subtracting the spectra of the reactants. Initial concentrations of gallic acid and catechol were 0.1 and 0.4 mM, respectively, at pH 3 and 0.01 M KCl ionic strength. Ratios listed are mol:mol of organic ligand:Fe(III). Reproduced with permission from ref. 31 (Copyright © 2013, American Chemical Society). | ||
The strong chelating abilities of Fe with organic ligands containing the catechol moiety and their redox chemistry are well established at the fundamental level, where the pH and the relative concentration of the metal to the organic ligand determine the nature of the results.122,123,127–129 More recently, this basic chemistry has been utilized in functionalizing surfaces and nanoparticles for applications in green chemistry130 and the development of biomedical and sensing devices.131–133
In the case of catechol, an intense green colour forms in the pH range 2–4 on mixing with Fe(III) as a result of the formation of a bidentate mononuclear catechol–Fe(II) complex with a ligand-to-metal charge transfer (LMCT) band around 700 nm.122,134 Complex formation between gallic acid and Fe(III) under acidic conditions (pH < 3) results in the formation of a blue colour with an LMCT around 660 nm and assigned to the bidentate mononuclear gallic acid–Fe(II) complex.124 Using electrospray ionization mass spectrometry, Ross et al.135 reported complex ion formation with the addition of Fe(III) to tannic ligands that included gallic acid. They determined that the formula [L–3H + Fe(II)]− could explain the observed masses. A more detailed experimental investigation using electrospray ionization mass spectrometry by Lutui et al.136 coupled with ab initio calculations also confirmed the oxidation state of iron in these complexes and proposed a number of different structures that could explain the results. Heyden et al.137 used X-ray based iron L-edge and carbon K-edge spectromicroscopy to analyse particles from oxic marine and freshwater sites for iron speciation and associations with organic matter. Fig. 7 summarizes the results of their paper by showing the types of organic functional groups and their distribution among the different iron-containing samples. The ubiquity of Fe(II) in these colloidal samples sheds some light onto the changes in the kinetics of redox cycling as a result of complexation to organic ligands.
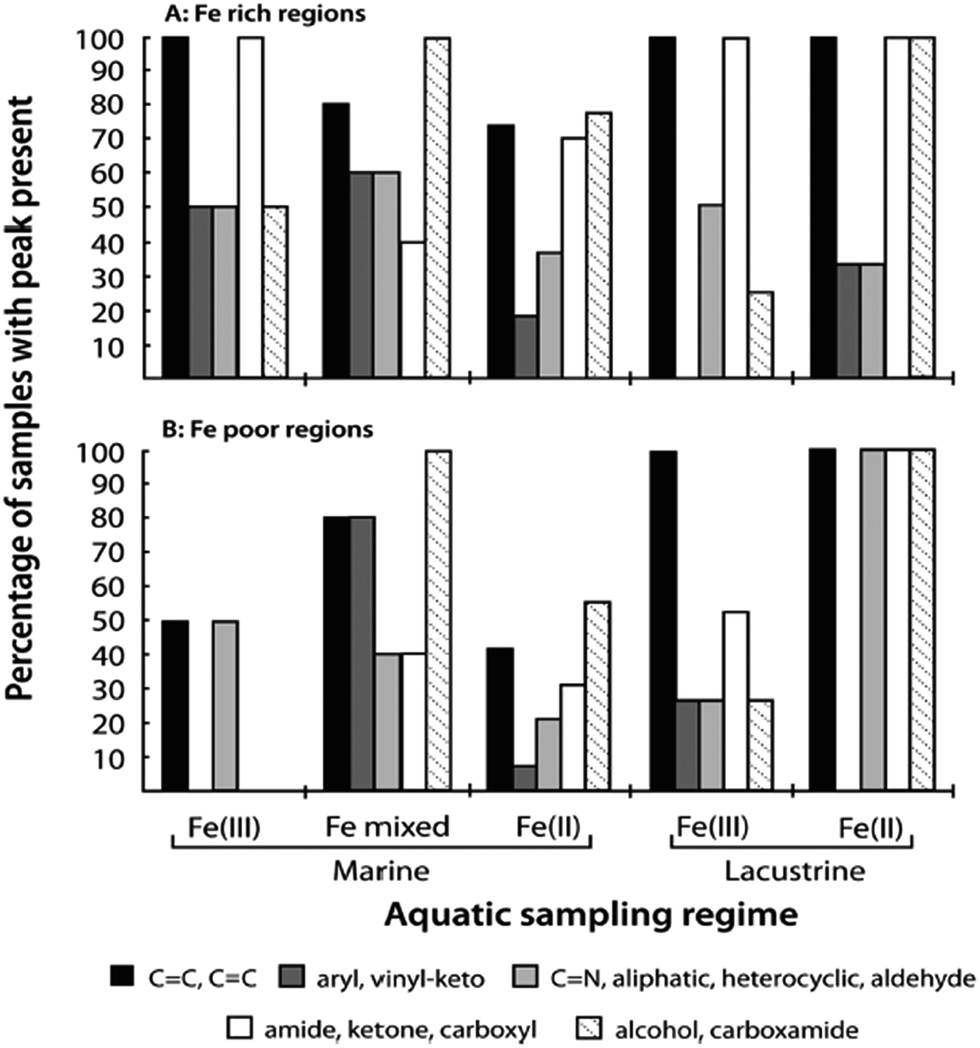 | ||
| Fig. 7 (A) Frequency of each set of organic functional groups as found in association with Fe-rich particulates of varying chemistry and sampling location. (B) Corresponding analyses of organic functional group frequency for Fe-poor regions of each particle or sample window. Reproduced with permission from ref. 137 (Copyright © 2014, American Chemical Society). | ||
The presence of the –OCH3 group as a substituent on the benzene ring, as in the case of guaiacol, favours the formation of oligomers at the expense of stable guaiacol–Fe complexes.138 The addition of iron results in the development of soluble amber-coloured (about 412 and 470 nm) dimers and trimers of guaiacol. These have also been observed by Hwang et al.139 from the biochemical oxidation of guaiacol by manganese peroxidase in the presence of H2O2. The presence of oxidants results in the formation of phenoxy radicals that initiate C–C coupling reactions to form dimers. Analysis of the UV-visible spectra of mixtures of solutions showed a decrease in the intensity of the bands in the 400–500 nm range on the overnight storage of solutions.139,140 Schmalzl et al.138 assigned the 470 nm peak to an unstable 4,4′-diphenoquinone intermediate to explain these observations. These molecular level results provide insights into the increase in optical properties observed experimentally from the interactions of Fe(III)141 and other metals142 with dissolved organic matter. In summary, the bulk aqueous phase chemistry of iron is dependent on pH, with different soluble species capable of chelating organic compounds with different affinities. Under dark conditions and with certain redox-active organic compounds, the cycling of Fe(II) and Fe(III) and the role of dissolved oxygen and other oxidants become important factors in determining the fate of organic compounds.
4. Surface chemistry of iron in the dark
4.1. Nature of surface water at different interfaces
Studies that highlight the molecular level differences between bulk and interfacial environments in atmospherically relevant systems have been the subject of thematic special issues of the Journal of Physical Chemistry C143 and Physical Chemistry Chemical Physics.144 In addition, the nature of surface water and its role in the heterogeneous reactions of sea salt and mineral dust particles with gas phase species such as ˙OH, O3, SO2, ˙NO2, HNO3 (to name a few) have been reviewed by Finlayson-Pitts17 and Grassian,145 respectively. The studies highlighted in these reviews showed that: (a) the liquid layer on the surface of sea salt is enhanced with ions (Cl−, Br− and I−) even at low RH, which directly affects the reaction probabilities of gases and interfacial acidity; (b) the surface versus bulk partitioning of ions such as nitrate in the presence of water depends on the amount of water; (c) surface water enhances ionic mobility, which leads to the regeneration of surface sites; (d) surface water on airborne dust and organic monolayers146 is “structured” compared with bulk liquid water, which affects their ability to act as efficient condensation nuclei; and (e) the photolysis of chromophores such as deliquesced surface nitrate is more efficient in producing gas phase O(3P), ˙OH and ˙NO2 than photolysis in solution. Grassian and co-workers147,148 reviewed studies on the heterogeneous chemistry of metal oxide and carbonate interfaces and how surface water plays a role in: (a) altering reaction pathways and surface speciation; (b) enhancing surface reactivity as a result of an enhancement in ionic dissociation and mobility; (c) solvating ions from the adsorbed phase; (d) inhibiting surface reactivity due to site blocking; and (e) hydrolysing reactants, intermediates and products.To investigate the role of organic compounds, the adsorption of water from the gas phase on flat surfaces terminated with organic functional groups was studied using IR spectroscopy, as reviewed by Moussa et al.146 and Asay et al.149 Analysis of the stretching mode of OH groups, v(OH), provides further insight into the nature of the hydrogen bonding network for surface water in different samples. For example, the v(OH) region in the IR spectrum of liquid water is characterized by a broad feature at about 3400 cm−1, which red shifts to about 3200 cm−1 in ice as a result of stronger and more ordered hydrogen bonds.150,151 It was found from experimental data and molecular dynamic (MD) simulations of water clusters on hydrophobic organic monolayers that the relative intensities of the 3200/3400 cm−1 components correlated with the number of hydrogen bonds among water molecules.146 Fig. 8 shows the adsorbed water as a function of RH for different surfaces with and without organic species. Water molecules in contact with organic monolayers at low RH have fewer hydrogen bonds and give rise to spectral components at 3200 cm−1, whereas molecules in the interior of water clusters have three and four hydrogen bonds similar to bulk water. In a related study, Nichols et al.152 studied the reaction of a benzophenone–catechol mixture with NO2 under dark conditions at 20% RH using ATR-FTIR and noticed an increase in the absorption of a broad feature between 2000 and 3000 cm−1 with the reaction time. This structured feature, with resolved components at 3100 and 2900 cm−1, was assigned to the v(OH) of strongly hydrogen bonded adsorbed water on the solid phase of more hygroscopic reaction products (mostly 4-nitrobenzene-1,2-diol).
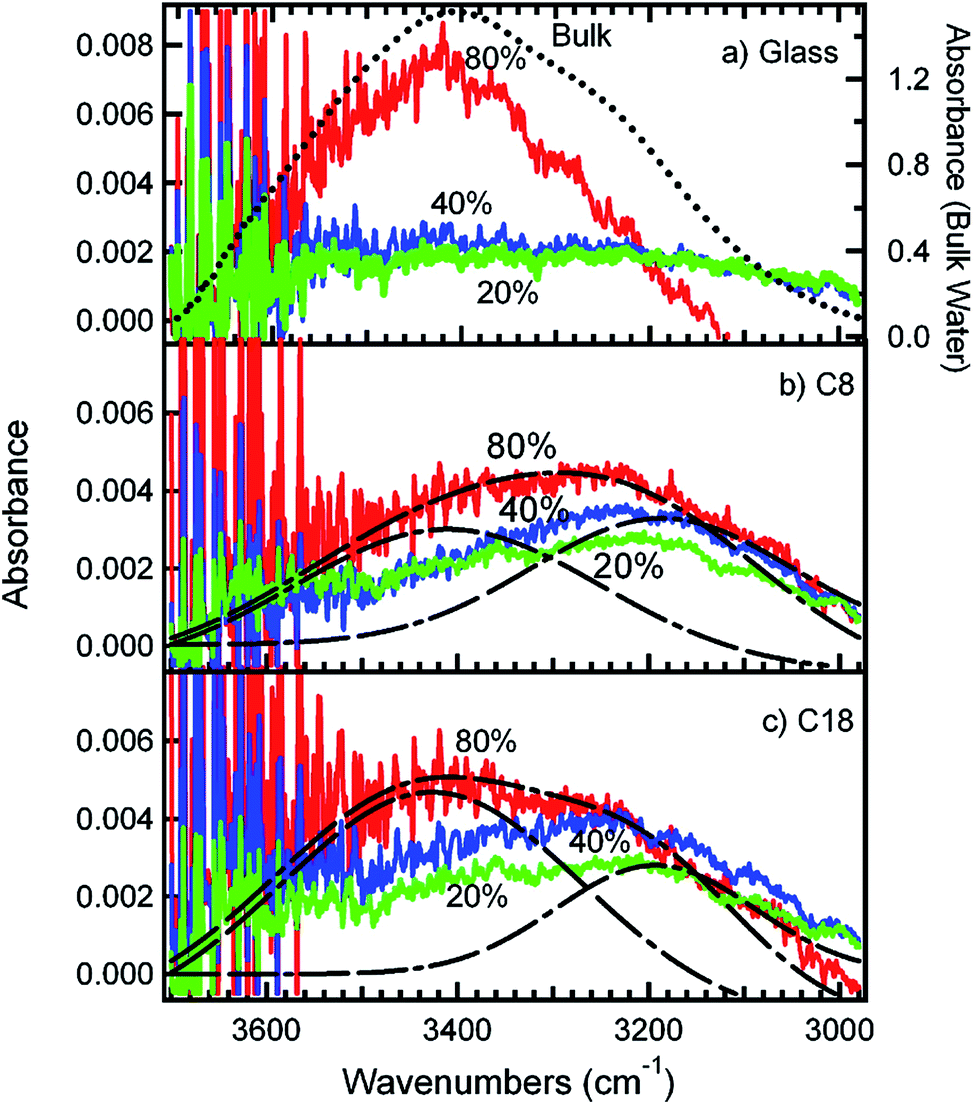 | ||
| Fig. 8 Infrared spectra of adsorbed water in equilibrium with water vapour at different percentages of RH on (a) plasma-cleaned glass, (b) glass coated with a C8 SAM and (c) glass coated with a C18 SAM. The dotted line in (a) is the spectrum of bulk liquid water. The dashed lines in (b) and (c) represent the fitting of the 80% RH spectra into two peaks centred at 3200 and 3400 cm−1, respectively. The different colours correspond to adsorbed water at different percentages of RH: green (20% RH), blue (40% RH) and red (80% RH). Reproduced with permission from ref. 146 (Copyright © 2009, American Chemical Society). | ||
Using diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), Cowen and Al-Abadleh36 recorded the IR spectra of water adsorbed on tannic acid powder as a function of RH before and after irradiation. Fig. 9 shows the difference absorbance spectra obtained using dry tannic acid as the reference. The spectral region between 1800–1000 cm−1 contains fundamental vibrations of organic functional groups and the bending mode of water, δ(H2O). Changes in this region due to water adsorption on organic surfaces are rarely analyzed (see references in Moussa et al.146). The trends observed in the spectral region below 1800 cm−1 suggest changes to the phase of the starting material with increasing RH as a result of the dissolution of tannic acid at the interface or the formation of tannic acid hydrates. Detailed analysis36 of the v(OH) region suggested the formation of a strong hydrogen bonding network in tannic acid hydrates arising from tannic acid–water or water–water interactions at the interface, which dominated at low and high RH, respectively. In addition, changes to the functional groups of tannic acid after irradiation seemed to influence the hydrogen bonding network in adsorbed water, as evidenced by the red shift of the broad feature maximum by about 39 cm−1 and the increase in the intensity of a lower frequency component at 3039 cm−1.
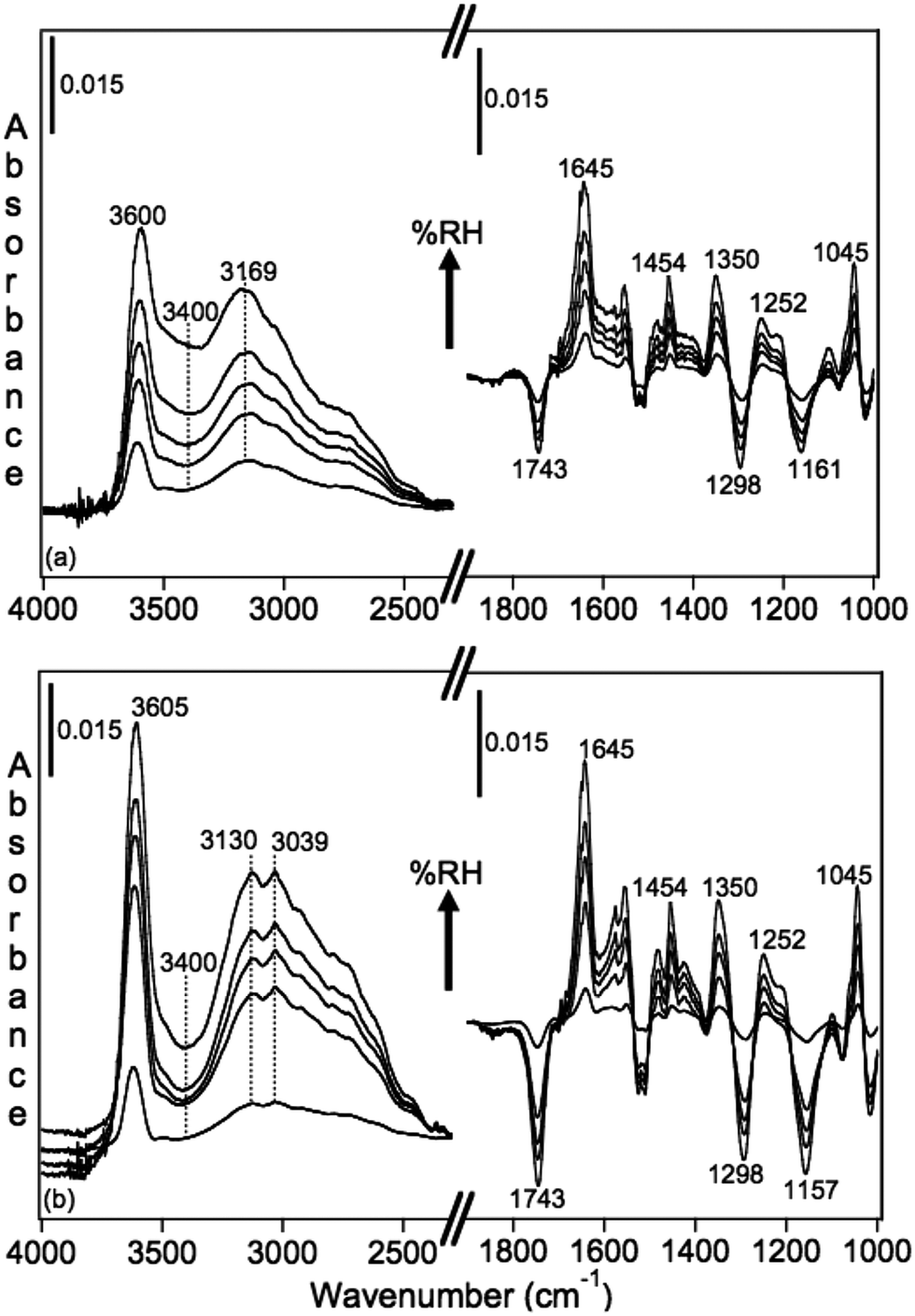 | ||
| Fig. 9 Absorbance spectra of water adsorbed on tannic acid as a function of increasing RH. (a) Unirradiated (5.5, 22, 40, 60 and 84% RH), and (b) after irradiation for 3 h at 20% RH (Xe lamp, 20 W m−2) at 2, 23, 40, 58 and 81% RH. Reference spectrum is tannic acid dried by flowing <1% RH air overnight. Reproduced from ref. 36. | ||
Using non-linear sum frequency generation (SFG), the spectra of water molecules at the vapour–water and organic–water interfaces were also studied.153,154 The SFG spectra of water at these interfaces show broad features extending from 3500 to 3000 cm−1, with a maximum around 3480 cm−1 for the vapour–water interface that blue shifts to about 3500 for the CCl4–water interface. This broad feature was assigned to water molecules involved in strong hydrogen bonding with nearby water molecules at the interface.153 High-frequency features have been observed in the SFG spectra of interfacial water molecules near 3700 cm−1, which are referred to as the “free OH” mode of highly oriented water molecules.153 This feature is narrow and centred at 3705 cm−1 for vapour–water that is red shifted to 3674 cm−1 for alkanes–water, 3669 cm−1 for CCl4–water and 3650 cm−1 for CDCl3–water interfaces. This sharp feature was used as an indicator for the presence of water–organic species interactions and the relative strength of this interaction: the lower the frequency of “free” v(OH), the stronger the water–organic species interaction. Hence the aforementioned trend observed in SFG studies is indicative of a weaker alkane–water interaction than the CCl4–water interaction and the strongest interaction is observed for water at the CDCl3–water interface as a result of its higher polarity. It was concluded from these studies, which were complemented by MD calculations, that these weak interactions drive a molecular ordering behaviour that extends well into the organic phase.153
We have studied the uptake of water on FeCl3 particles as a function of the RH using DRIFTS.31 This is a hygroscopic salt that has been reported to be completely deliquesced at 77% RH for suspended micron-sized particles,155 which is a much higher value than that estimated from the bulk solubility (45%) by Cohen et al.155 Our IR data showed that at RH values < 40% the v(OH) band had maxima at 3572 and 3070 cm−1, which became enveloped by features at 3375 and 3228 cm−1 with increasing RH. The presence of OH groups with fewer hydrogen bonds than those found in the bulk phases was manifested by spectral features with frequencies between 3700 and 3500 cm−1. The data suggested that water–FeCl3 interactions at low RH resulted in the formation of only a few, but strong, hydrogen bonds and that, with increasing RH, the water–water interactions became more pronounced and gave rise to a more “liquid-like” hydrogen bonding network. Interestingly, the uptake of water on FeCl3 particles in our studies did not exhibit a sharp or well-defined increase at deliquescence. This behaviour was observed previously in electrodynamic balance experiments155 and was explained by the formation of polymeric ferric hydroxide species with increasing water uptake according to:
| mFe3+(aq) + nH2O(l) → [Fem(OH)n](3−n)+(aq) + nH+(aq) | (8) |
We also investigated the effect of irradiation on the hydrogen bonding network of water adsorbed on FeCl3 at 30% RH.30 Fig. 10 shows the changes in δ(H2O) as a function of irradiation time from difference spectra with growing features at 1643 and 1601 cm−1, suggesting that the iron species that formed with irradiation were [Fe(H2O)6]2+, where a sixth water molecule bound directly to Fe(II) to complete its first hydration shell to six ligands.162,163 The v(OH) region appeared to be narrower than that observed in the dark, with additional narrow peaks on the low frequency shoulder at 3221 and 3182 cm−1. This was a manifestation of the disturbances to the hydrogen bonding network in the first and subsequent shells around the Fe(III) and Fe(II) centres as a result of the absorption of light.
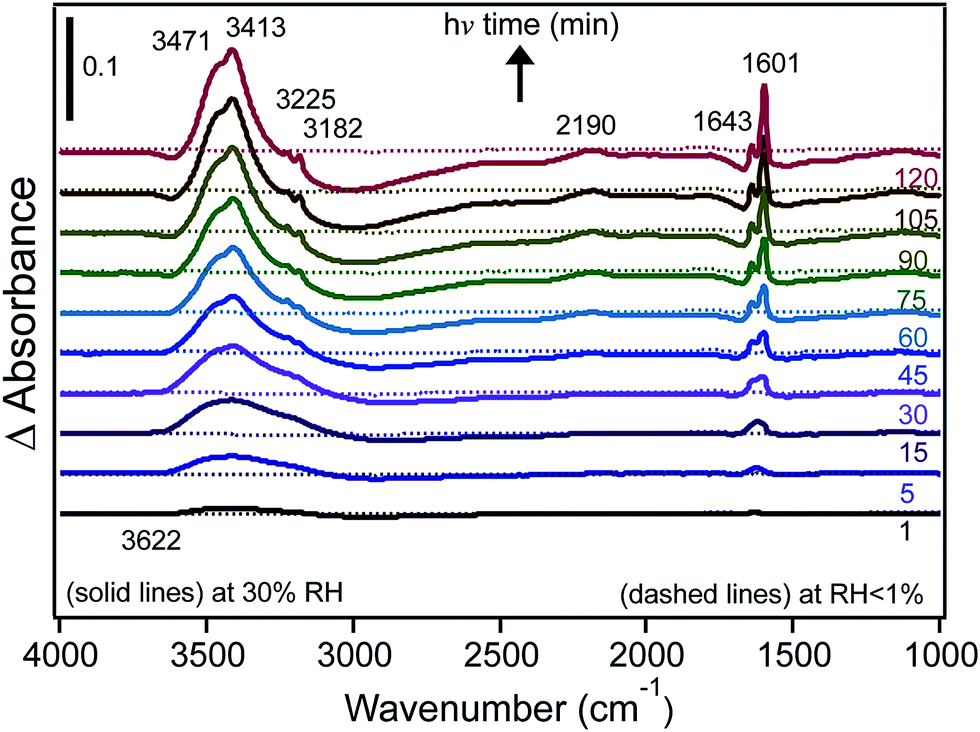 | ||
| Fig. 10 Difference DRIFTS absorbance spectra collected at 30% RH (solid lines) and under dry conditions (dashed lines) as a function of irradiation for a sample containing 1% FeCl3 (w/w) in diamond powder in the absence of adsorbed catechol (as a control experiment). Reprinted with permission from ref. 30 (Copyright © 2014, American Chemical Society). | ||
The structure of water adsorbed on γ-Fe2O3,156 nano Fe2O3 (ref. 157) and goethite particles158–160 was studied using infrared spectroscopy as a function of RH. Spectra collected by Goodman et al.156 showed the growth of the δ(H2O) band centred at 1640 cm−1, which was similar to spectra collected for other metal oxides. With increasing RH, the v(OH) absorption band split into two bands with centres at 3589 and 3113 cm−1, which were assigned to γ-FeOOH surface groups. The characteristic signature of the surface hydroxyl groups on Fe2O3 was investigated by Baltrusaitis and Grassian157 by performing isotope experiments using D2O(g) and H2O(18) vapour. The absorption bands associated with surface O–H groups were similar using both H2O and H2O(18) vapour (around 3687, 3655 and 3365 cm−1), whereas the bands associated with surface O–D groups were seen at 2712, 2697 and 2510 cm−1. This study also examined the adsorption of CO2(g) on Fe2O3 particles in the presence of adsorbed water and provided data for the formation of surface bicarbonate (FeOCO2H), which, in the presence FeOH groups, formed FeCO3− and adsorbed H3O+, thus acidifying the surface. A related study was conducted by Song and Boily159 to identify the types of hydroxyl groups on goethite nano-rods under dry conditions and with increasing RH. Fig. 11 summarizes their findings on dry particles where the experimental IR spectrum was correlated with a calculated spectrum using MD calculations. On the adsorption of water on this hydroxylated surface of goethite, the high-frequency absorption band decreased in intensity and new features around 3582, 3551 and 3510 cm−1 appeared below 10 Torr water vapour (<50% RH). These were enveloped by the most intense band around 3340 cm−1, leading to the interpretation that liquid-like water thin films formed at the goethite surface. Wijenayaka et al.158 investigated the effect of particle size on the adsorption of water on goethite nano- and micro-rods and found that the total amounts of water normalized to the surface area were similar. A size effect was observed in the adsorption of nitric acid to these materials, in which the micro-rods took up more irreversibly bonded HNO3 than the nano-rods. This was explained by surface structural changes to the hydroxyl groups; a decrease in the total reactive hydroxyl group density per unit area was observed in going from larger to smaller goethite particles.158
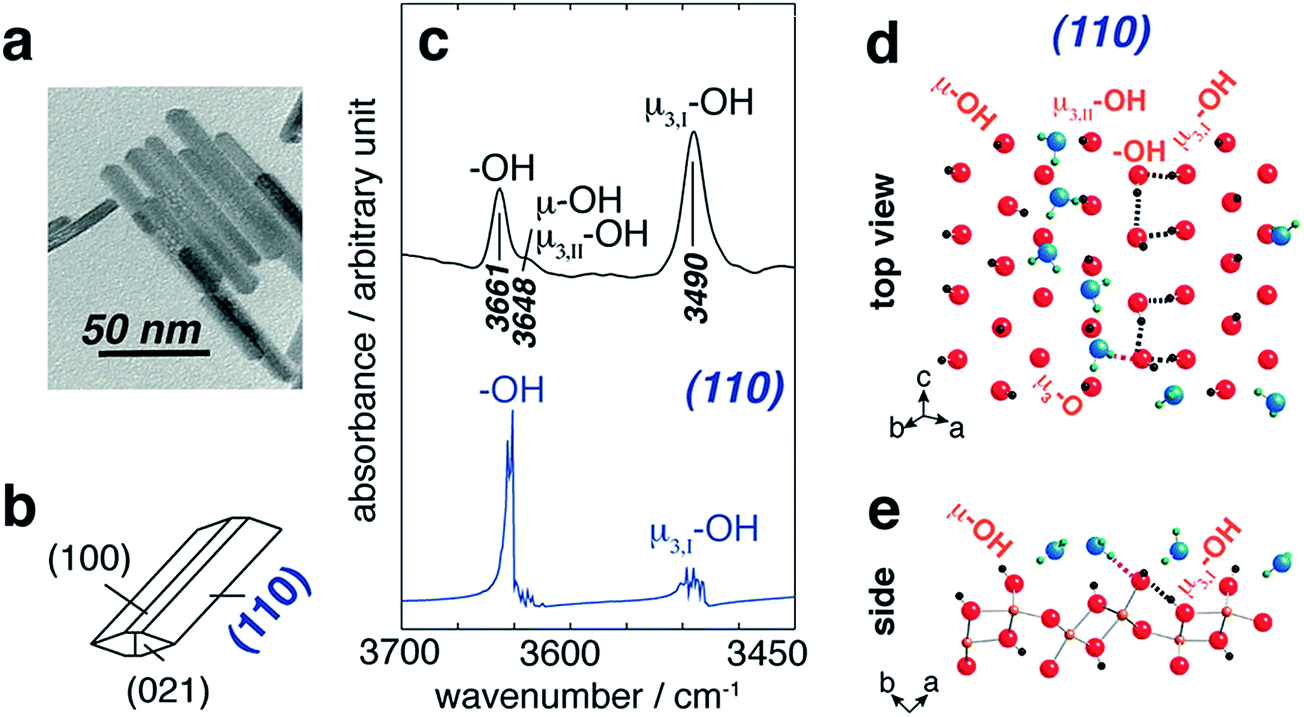 | ||
| Fig. 11 (a) TEM image of goethite particles (scale bar 50 nm) and (b) idealized particle morphology. ((c), top) FTIR band assignments of hydroxyl groups on goethite surfaces and ((c), bottom) the MD-derived power spectrum of the (110) plane. Schematic representation of the first-layer waters adsorbed at the (110) surface of goethite from the top view (d) and a side view (e) as obtained from a snapshot of an MD simulation. Black dashed lines represent original H-bonding patterns in water-free systems, whereas the pink lines represent new H-bonds in the presence of water. Reprinted with permission from ref. 159 (Copyright © 2013, American Chemical Society). | ||
To summarize this section, gas phase water interactions with solid or liquid organic surfaces, iron salts and iron (oxyhydr)oxides lead to the formation of an adsorbed phase with a different hydrogen bonding network to that of water in the bulk liquid and solid phases. Depending on the hygroscopicity of the solid material, the surface water can either increase the ionic mobility with impacts on the surface acidity or increase the degree of hydroxylation of the surface sites. Each case presents a unique chemical environment for reactions with organic compounds.
4.2. Reactivity of surface iron with organic compounds: complexation and enhanced dissolution
Soluble and insoluble iron-containing materials are reactive towards organic compounds because iron centres are strong complexing agents and, in the presence of oxidants such as H2O2, they initiate Fenton and Fenton-like reactions. The complexation of organic compounds to iron could affect their degradation in the dark and their photochemical decay rates depending on the amount of surface water. This is because the hydrogen bonding network among water molecules at the gas–solid interface is different from that in bulk water. In many cases, this chemical reactivity leads to the dissolution of iron (oxhydr)oxides, with consequences for the bioavailability of iron and the chemical and photochemical reactivity of the system. In this section, several examples related to this topic are highlighted.The heterogeneous chemistry of soluble iron salts such as FeCl3 is relevant to that of aged iron-containing mineral dust particles, whereas that using hematite mimics the chemistry of freshly emitted dust. The adsorption of short-chain volatile organic compounds (VOCs), including aliphatic alcohols, ketones and acids, on a number of minerals and metal oxides has been reviewed by Usher et al.90 In addition, the catalytic activity of natural and synthetic iron (oxyhydr)oxides in Fenton and Fenton-like reactions has been reviewed by Pereira et al.164 A less studied topic is the adsorption of aromatic VOCs on solid metal-containing salts.
We recently investigated the complexation of catechol to FeCl3 particles under dry and humid conditions using DRIFTS.31 The spectra of surface species shown in Fig. 12A resemble those collected for aqueous phase catechol in the absence of Fe(III) in solution at acidic pH values. The spectral data suggest that, under dry conditions, catechol adsorbs molecularly and is fully protonated, showing no evidence for complexation with Fe(III). On increasing the RH until it reached 30%, the spectra collected as a function of time (Fig. 12B) showed clear changes to the shape of the bands and the intensity assigned to the functional groups in catechol, in addition to an increase in the amount of surface water (band at 1620 cm−1). The enhancement in Fe(III) mobility under these conditions led to the formation of stable catechol–Fe complexes at the gas–solid interface. These spectra resembled that collected for a catechol–Fe solution at pH 4, with a few peaks slightly shifted as a result of the unique hydration environment at the gas–solid interface. These data also showed that the uptake of water by hygroscopic FeCl3 particles changed the interfacial pH to acidic values, giving rise to a different complexation behaviour with organic compounds from that of coordinated Fe(III) in hematite.
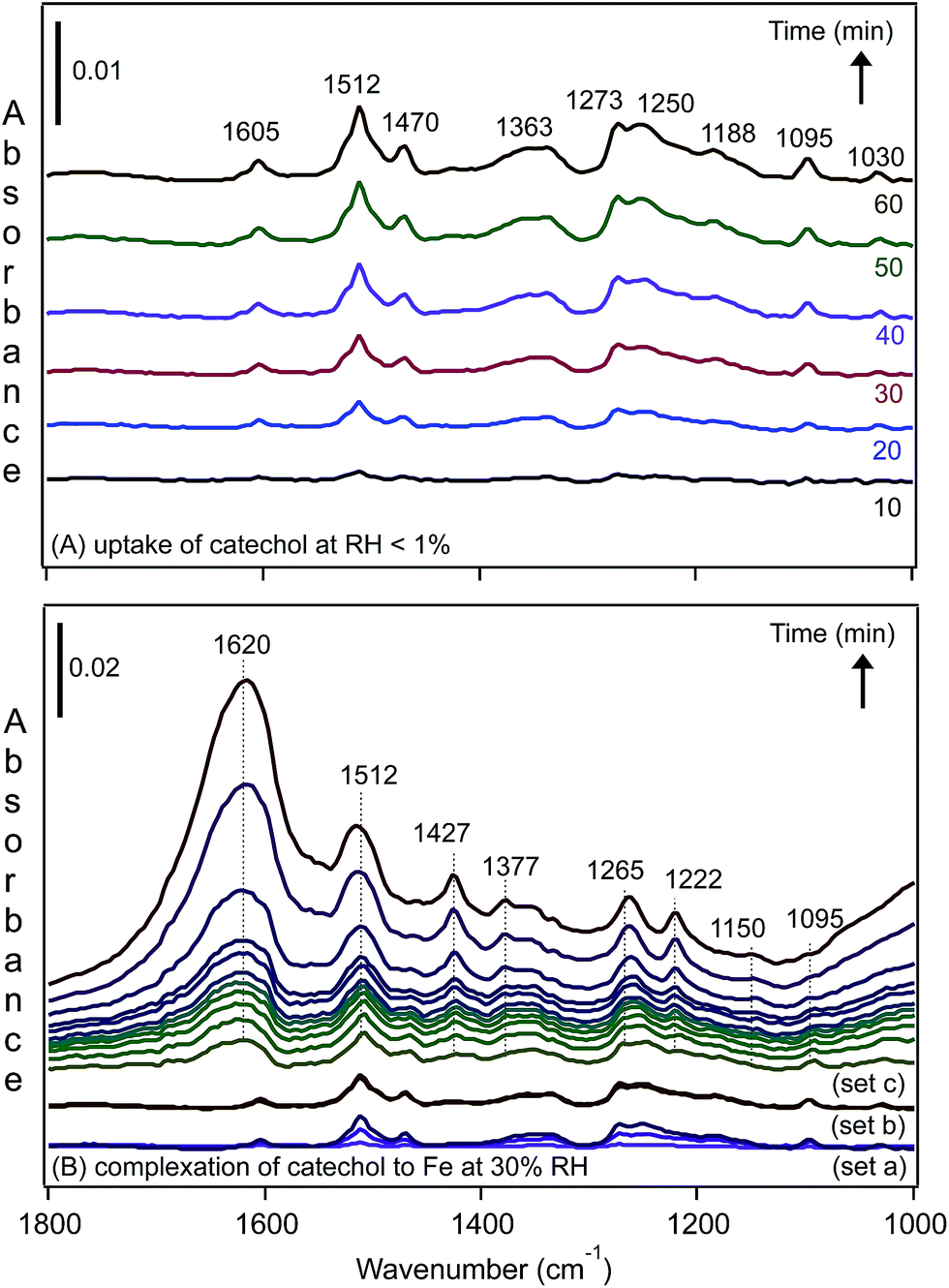 | ||
| Fig. 12 Representative time-dependent DRIFTS absorbance spectra of surface catechol on 1% FeCl3(s) as a result of (A) flowing gas phase catechol in the dark under dry conditions, RH < 1% and (B) increasing the RH to 30%, resulting in complexation to Fe(III) in the presence of adsorbed water. The spectra shown in set (a) are the same as shown in (A) at times of 10, 30 and 60 min while flowing gas phase catechol under dry conditions. The spectra shown in set (b) are for adsorbed catechol after stopping the flow of catechol vapour with a continuous flow of dry air for 10, 30 and 60 min. The spectra shown in set (c) are for adsorbed catechol as the RH of the air flow is increasing to 30% RH (no gas phase catechol) at times of 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 10, 30 and 60 min (from bottom). Reproduced with permission from ref. 31 (Copyright © 2013, American Chemical Society). | ||
Insoluble iron-containing materials include naturally abundant and industrially important iron (oxyhydr)oxides such as hematite, goethite and ferrihydrite.164–166 To show the contrast of the described studies with hematite, we conducted similar experiments for catechol vapour uptake under dry and humid conditions. Because hematite is an insoluble metal oxide, catechol adsorption occurs via exchange with ligands on the Fe sites, as shown in reaction (9):
2![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) FeOH + catechol → [Fe–catechol–Fe] + 2H2O FeOH + catechol → [Fe–catechol–Fe] + 2H2O
| (9) |
The release of water caused little change to the acidity of the adsorbed water layer.
Fig. 13 shows the spectral features of surface catechol under dry and humid conditions. When compared with the spectra of aqueous catechol,31 the data under the dry conditions suggested the formation of monodentate catechol–Fe complexes that were hydrogen bonded to neighbouring sites. For the spectra collected at 30% RH, the features assigned to the surface catechol closely resembled those observed for the adsorption of aqueous phase catechol on hematite167 and goethite168 particles at neutral to basic pH values. So, again, with this comparison of the aqueous phase spectra collected at known pH, we can obtain an idea about the interfacial pH in the absence of direct measurements.
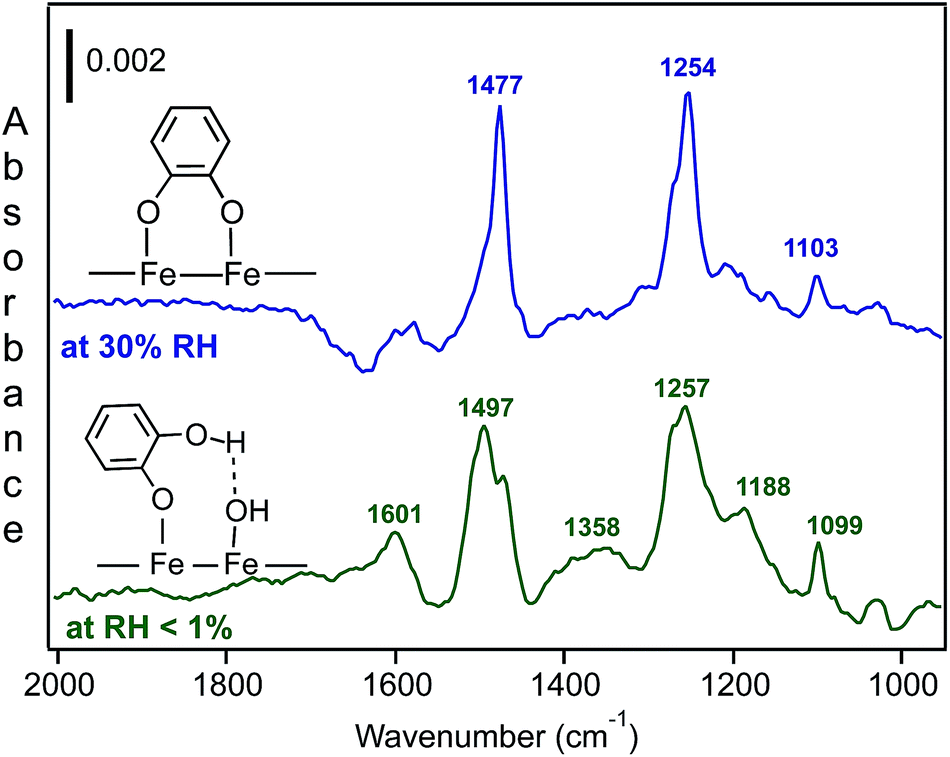 | ||
| Fig. 13 DRIFTS absorbance spectra of catechol vapour uptake on solid hematite nanoparticles (1% w/w in diamond powder) under dry (bottom, RH < 1%) and humid (top, 30% RH). The reference spectrum used is that collected for hematite nanoparticles prior to introducing catechol vapour. Original work from experiments in the author's laboratory. | ||
Chemical and biological processes that lead to the dissolution of iron-containing materials under various conditions have been of interest to researchers because they affect the biogeochemical cycle of iron and other species.2,61,62,169–171 Examples of these processes from a recent review of the geochemical literature are shown in Fig. 14.172
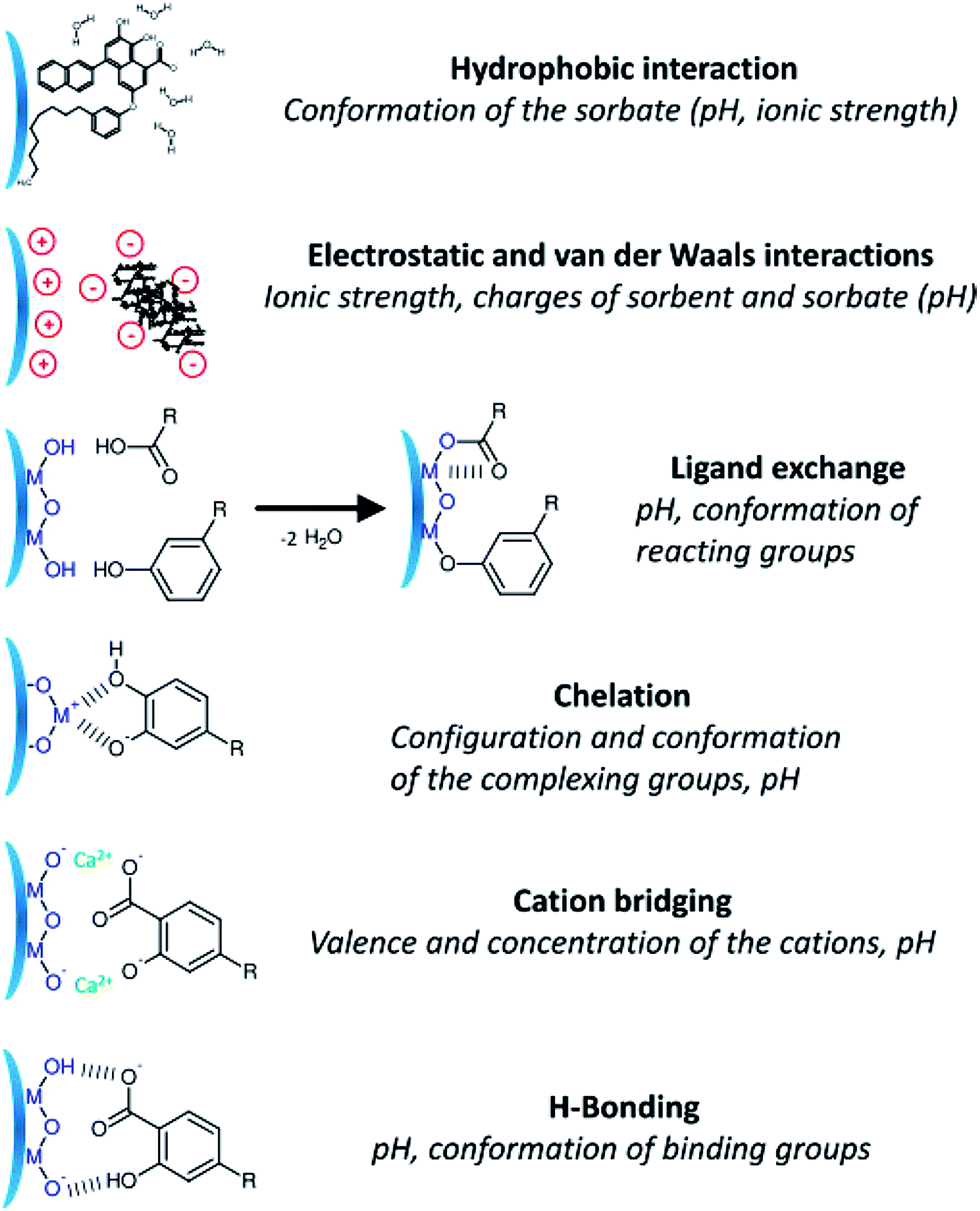 | ||
| Fig. 14 Schematic description showing the diverse mechanisms for the interaction of dissolved organic matter on the surface of metal oxides. Reproduced with permission from ref. 172 (Copyright © 2014, American Chemical Society). | ||
In another review, Liu et al.173 summarized studies of the adsorption of small molecular weight organic ligands and fulvic and humic acids on goethite surfaces. Cwiertny et al.174 synthesized the literature on the specific dissolution processes of iron (oxyhydr)oxides that included proton-promoted, ligand (oxalate)-promoted, dark reductive via ascorbic acid, and photochemical reductive dissolution in the presence of oxalate. The following section highlights selected studies on the role of organic molecules that mimic HULIS in the complexation and dissolution of iron (oxyhydr)oxides over a wide pH range.
Using surface-sensitive infrared spectroscopy, ATR-FTIR and batch dissolution experiments, Gulley-Stahl et al.167 correlated the IR spectra of catechol complexes on high surface area Fe2O3 particles and other metal oxides (Cr2O3, TiO2 and MnO2) with their dissolution behaviour (i.e. [metal]free) in the pH range 3–10. The results on Fe2O3 showed that dissolution after 30 min of mixing had a higher dependency on pH than on the ionic strength. The presence of catechol promoted dissolution under neutral and basic conditions compared with the proton-driven dissolution that was significant under acidic conditions (pH < 5). This was explained by the formation of inner sphere catechol complexes at pH > 5 with a higher degree than outer sphere complexes under acidic conditions. In the light of other studies that showed that bidentate binuclear complexes of catechol inhibit dissolution, these workers inferred that bidentate mononuclear complexes at pH > 5 were responsible for promoting dissolution.
In a later study, Yang et al.168 studied the adsorption of catechol on goethite particles using ATR-FTIR and complemented these measurements with density functional theory (DFT) calculations of catechol complexes in the mononuclear monodentate (M–M), binuclear bidentate (B–B) and mononuclear bidentate (M–B) forms (Fig. 15). By examining the time profile of spectral features assigned to these complexes, they concluded that M–M and B–B complexes coexisted in the pH range 5–9, with the possibility of partial conversion of M–M to B–B via proton exchange with neighbouring surface sites under basic conditions and high surface coverage. The dissolution of goethite was studied in the presence and absence of catechol for 12 h in the dark under neutral to basic pH, where it was found that dissolved iron concentrations were below the detection limit of 5 μg L−1. This finding was explained by the dominance of B–B complexes that inhibited dissolution under these conditions and the possibility of dissolution–re-adsorption as observed in oxalate–goethite systems. In another study by the same group,175 the adsorption of salicylate was investigated in the presence and absence of catechol using ATR-FTIR and DFT calculations under neutral to basic conditions. The data revealed the formation of M–M complexes through the carboxylic acid group. The addition of catechol resulted in the formation of M–M and B–B complexes, which coexisted with salicylate complexes. It was also found that catechol replaced some of surface salicylate with no enhancement in goethite dissolution.
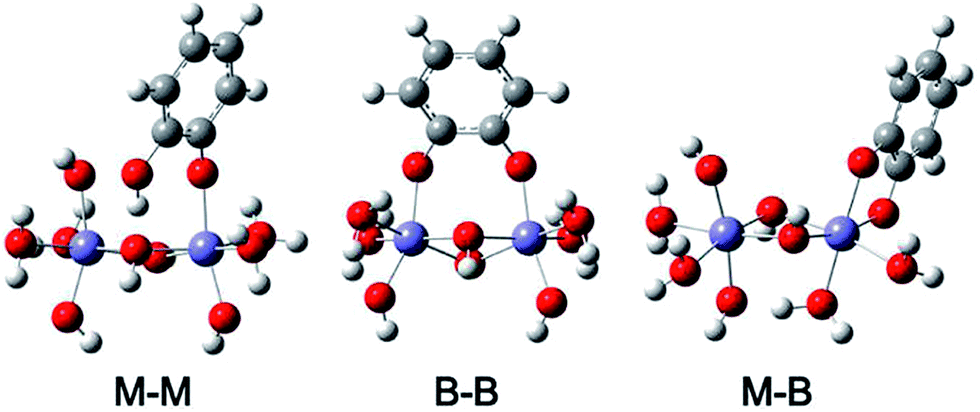 | ||
| Fig. 15 Optimized structures of catechol complexes on iron oxide clusters calculated using DFT-B3LYP with 6-31+G** basis set and IEFPCM solvation model (M–M), mononuclear monodentate; (B–B) binuclear bidentate; and (M–B) mononuclear bidentate. Reproduced with permission from ref. 168 (Copyright © 2012, American Chemical Society). | ||
Redox-active organic compounds promote the reductive dissolution of iron (oxyhydr)oxides. Anschutz and Penn176 investigated the reductive dissolution of crystalline ferrihydrite and goethite nanoparticles using hydroquinone as an electron donor. The production of p-benzoquinone and Fe(II) was monitored using HPLC and a ferrozine assay, respectively. The results showed that the surface area normalized rates were 100 times faster for ferrihydrite than for goethite particles, with faster rates observed for smaller sizes of nanoparticle. These results were explained by the lower degree of crystallinity and higher surface energy of ferrihydrite relative to the goethite nanoparticles. Because quinones play a key role in electron transport in minerals and microbes, Orsetti et al.177 reported measurements of the reduction potential of reactive iron species at the goethite surface using a non-sorbing quinone species, namely anthraquinone-2,6-disulfonate. Fig. 16 shows a comparison of the reduction potential of this system in relation to other environmentally relevant redox couples at pH 7.
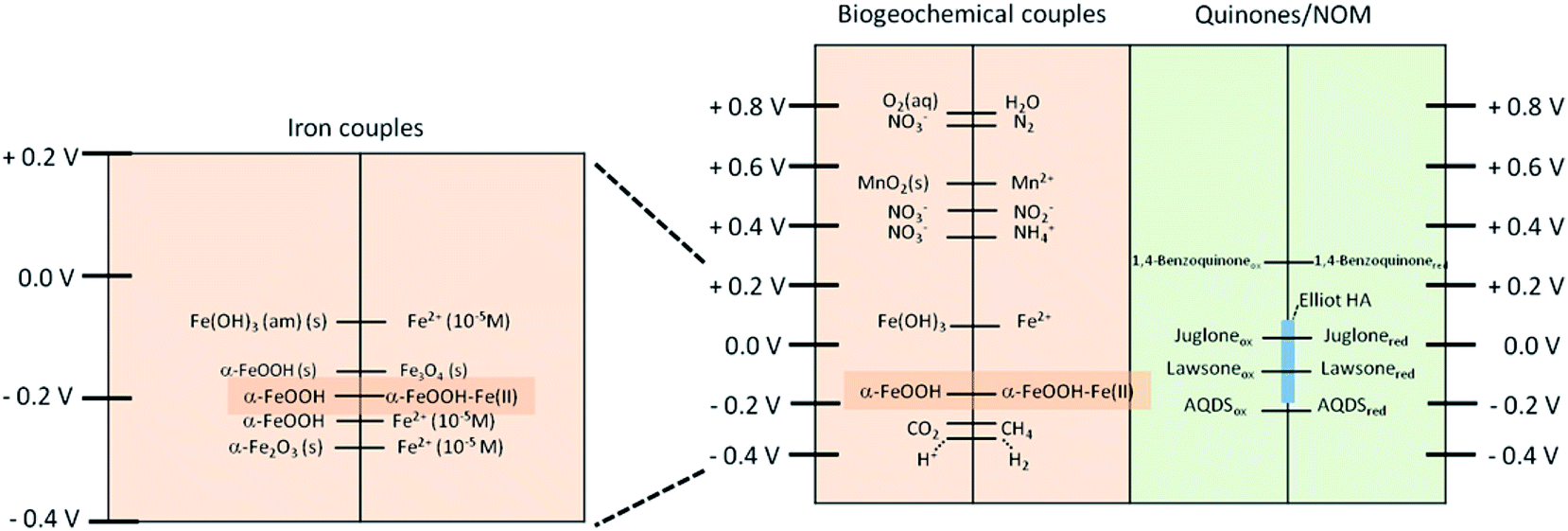 | ||
| Fig. 16 Comparison of the reduction potentials of some environmental relevant redox couples at pH 7 vs. the SHE (E′0H). Reprinted with permission from ref. 177 (Copyright © 2013, American Chemical Society). | ||
Shi et al.178 studied the reductive dissolution of goethite and hematite nanoparticles by reduced flavins, a class of organic compounds secreted by marine iron-reducing bacteria, Shewanella sp., with chemical structures that resemble HULIS. The results showed a higher reactivity for hematite than goethite in the pH range 4–7 under anaerobic conditions based on surface area normalized dissolution rates. When compared with poorly crystalline ferrihydrite, heterogeneous electron transfer from the reduced flavins was orders of magnitude higher than that using crystalline goethite. Other factors that affected the extent of reaction and the initial rates included the structure and functional groups of the organic substrate, the redox potential of the reductants and the iron (oxyhydr)oxides, the aggregation state and the pH.
To investigate the dominant mechanism that leads to the dissolution of iron in dust, Shi et al.179 collected iron-containing dust samples from two sites that represented sources of Saharan and Asian desert dust. In the laboratory, they simulated the effect of cycling between wet aerosols (i.e. acidic conditions) and cloud droplets (i.e. higher pH conditions) on the concentration of dissolved iron over a 3 h timeframe. The results showed that insoluble iron dissolved readily under the acidic conditions relevant to wet aerosols, whereas at higher pH values the dissolved iron precipitated as poorly crystalline nanoparticles. Therefore, in the long-range transport of mineral dust particles, the time spent as either a wet aerosol or in clouds will affect the amount of bioavailable iron on deposition. Acidic uptake within clouds can also enhance the dissolution of iron. As noted by DeMott et al.,180 cycling between the liquid, semi-solid and solid phases of aerosols takes place as result of changes in the temperature and RH. Variable solvent to solute ratios also change the pH and the relative importance of surface versus bulk chemistry.
In a related study by Chen and Grassian,181 the ligand- versus proton-promoted dark dissolution of iron by acids was investigated using coal fly ash and Arizona test dust as representatives of anthropogenic and natural mineral dust, respectively. Although the pH of the solutions was maintained at 2, the relative capacities of the three acids investigated to dissolve iron were in the order: oxalic acid > sulfuric acid > acetic acid. The formation of bidentate complexes of surface iron with oxalate explained the great extent of iron dissolution compared with monodentate complexes with acetate. It was noted that, at low oxalate concentrations, the competition of dissolved iron and surface iron for oxalate complexation suppressed the dissolution rate of iron. This study highlighted the similar role that could be played by other organic ligands that are preferentially associated with atmospheric dust aerosols, such as C2–C12 dicarboxylic acids, C16–C30 fatty acids182 and polycyclic aromatic hydrocarbons.183
The accelerated dissolution of iron (oxyhydr)oxides was observed when these materials were trapped in ice in the presence of organic electron donors in the dark.184 Fig. 17 shows the concentration of total dissolved iron, [Fetot], from the dissolution of goethite and maghemite in the bulk aqueous phase and in ice samples in the presence of organic ligands under acidic conditions. The authors found that most of the dissolved iron was in the ferric form, suggesting that this dark dissolution process was not reductive. The highest rates of dissolution were found with high surface area iron oxides and strong iron-binding ligands. The results were explained by a “freeze concentration effect” where solid particles, organic ligands and protons were concentrated in the liquid-like ice grain boundary region. Fig. 18 shows the location of the grain boundary region in ice crystals.185 There have been a number of reviews on the structure of the surface of snow186 and on other types of chemical processes that take place there.187
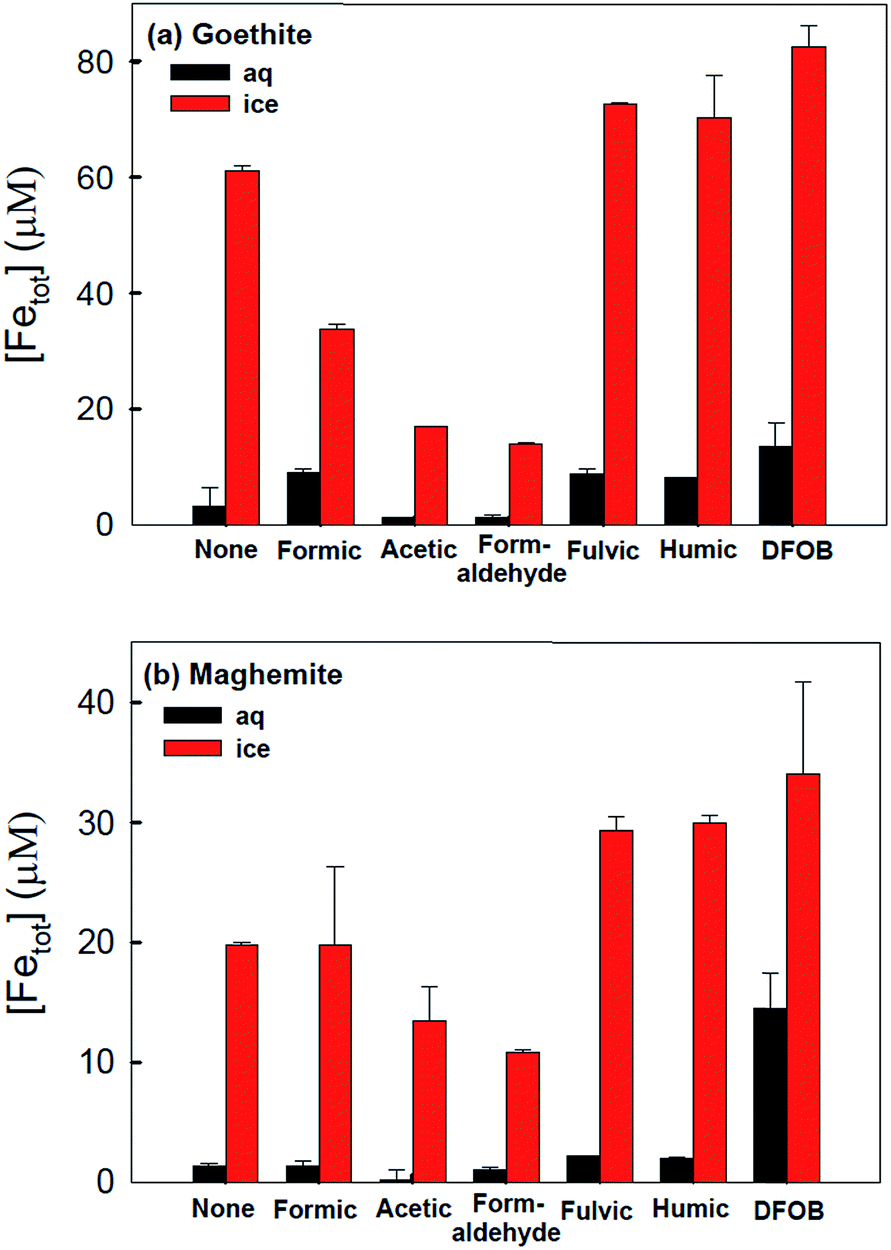 | ||
| Fig. 17 Production of total dissolved iron from (a) goethite (α-FeOOH) and (b) maghemite (γ-Fe2O3) under dark conditions in the presence of various organic complexing ligands in water (25 °C) and ice (−20 °C) after 72 h. Experimental conditions: [α-FeOOH or γ-Fe2O3]i = 0.2 g L−1, [organic ligand] = 6 mM, [fulvic or humic acid] = 1 ppm, [desferrioxamine B (DFOB)] = 50 μM, and pHi = 3.00 ± 0.05. Reproduced with permission from ref. 184. | ||
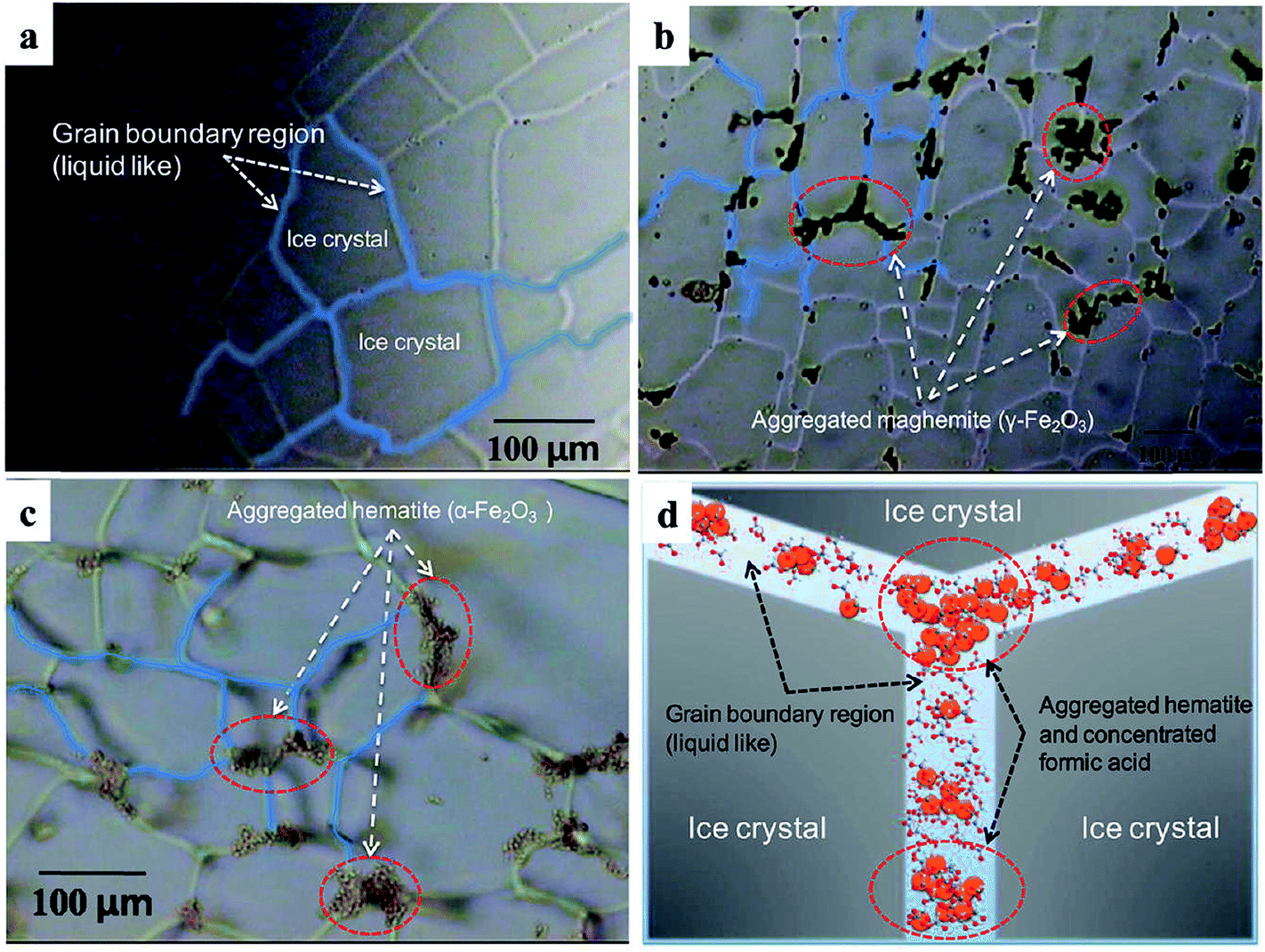 | ||
| Fig. 18 Optical images of polycrystalline ice showing the highly aggregated iron oxide particles trapped in ice veins (grain boundary region) at −20 °C. (a) Pure ice, (b) ice with maghemite (γ-Fe2O3) (1 g L−1), (c) ice with colloidal haematite (0.8 g L−1) and (d) schematic illustration of concentrated hematite particles (orange circles) and formic acids (ball-and-stick models) in ice grain boundary region. Reproduced with permission from ref. 85 (Copyright © 2010, American Chemical Society). | ||
To gain an insight into the mechanisms of the size-dependent dissolution rates of iron (oxyhydr)oxides, Lanzl et al.174 studied the influence of the primary particle size and solution pH (from acidic to approximately neutral) on 8 and 40 nm hematite nanoparticle aggregates. The smaller particles exhibited a 3–10 times enhancement in the mass-normalized reactivity from the pH dependent dissolution rates. This study also investigated the influence of aggregation on the dissolution of hematite, which led to a loss of reactive surface area as previously observed in the dissolution of goethite nano-rods.188 They found that the dissolution of aggregated 8 nm particles was uniform, whereas preferential etching at the edges and structural defects took place for 40 nm particles. Although this study and an earlier one by Cwiertny et al.189 on the dissolution of goethite nano- and micro-rods were conducted using oxalic and ascorbic acids as the organic ligands, they served as blueprints for future dissolution rate experiments using organic ligands other than chelated iron.
This section has summarized key investigations on the dark complexation of organic ligands from the gas and liquid phases to surface iron in soluble and insoluble materials, how this process influences the cycling between Fe(II) and Fe(III) species and the overall dissolution of iron oxides. These results provide the first step in our mechanistic understanding of the extent of photochemical reactions driven by iron under various reaction conditions, as detailed in the following sections.
5. Bulk photochemistry of iron
Studies related to the photochemistry of transition metals and chromophores relevant to aqueous aerosols have been the subject of a number of reviews and book chapters.13,14,16–19,190 The kinetics and mechanisms of these photochemical reactions are pH dependent as a result of speciation.100 Fig. 19 shows the cross section of Fe(III) species a function of pH, which overlaps with the solar actinic flux at λ > 290 nm.1,190,191 When compared with similar data for important chromophores in atmospheric aerosols (nitrate, nitrite and HULIS)14 and also H2O2,192 the cross section values in Fig. 19 are nearly 100 times larger than those of these chromophores. The complexation of Fe(III) with organic compounds results in a red shift in its UV absorption spectra to the visible region of the electromagnetic spectrum, with consequences for the overall photochemical reactivity of the system.31,124,129,141,193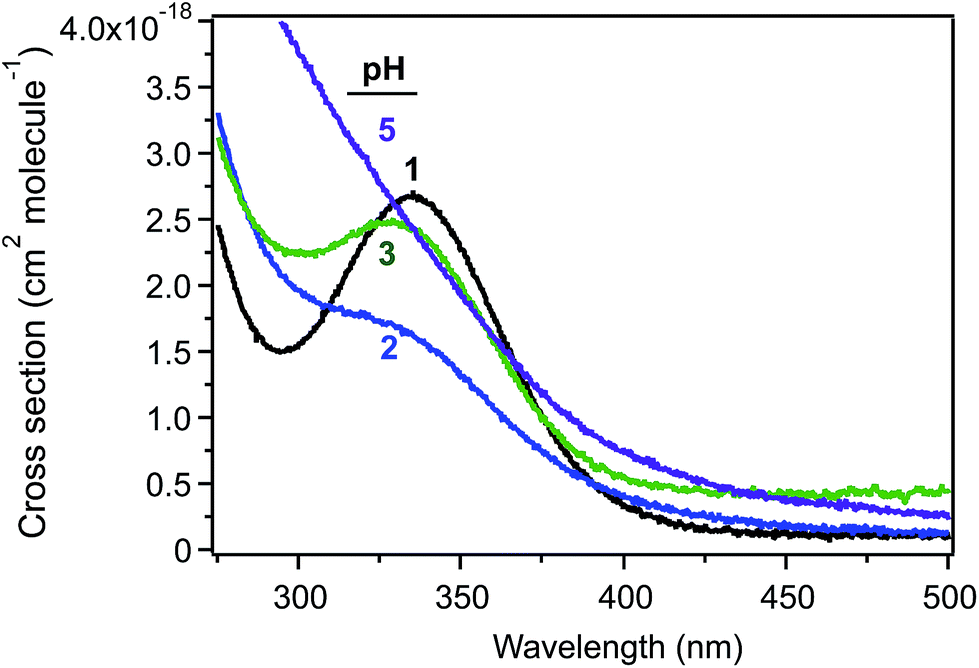 | ||
| Fig. 19 Cross section of Fe(III) species as a function of pH using 1 mM FeCl3 solution. | ||
The photochemistry of Fe(III) species in aqueous aerosols provides an important pathway for photodegrading WSOC.16 Reactions involving aqueous Fe(III) can be described by eqn (10) and (11):
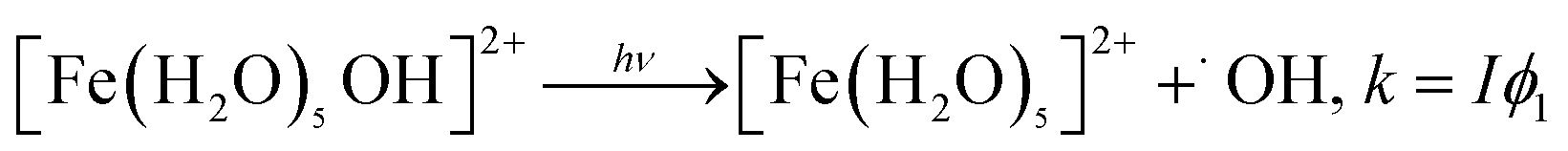 | (10) |
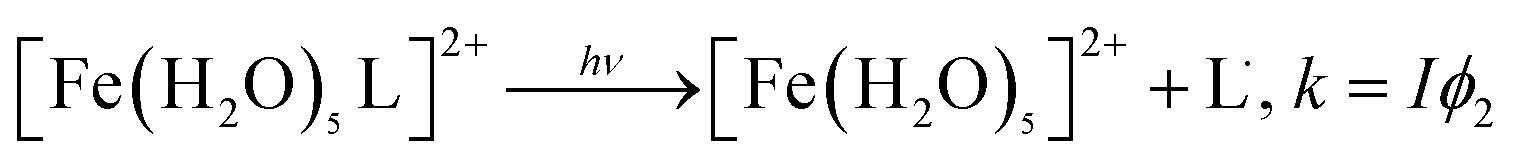 | (11) |
5.1. Iron as a photosensitizer in the degradation of WSOC
Interest in the iron-driven aqueous phase photosensitized degradation of organic compounds is driven mainly by the desire to treat wastewater contaminated by soluble natural organic matter and phenolic compounds produced from the decay of plants or the food and pigment industries.102,103,105 This chemistry can also be used to explain homogeneous reactions in cloud droplets41 and atmospheric organic aerosols.106 In this photochemistry, Fenton reagents, such as Fe(II) and H2O2, are most commonly used as efficient producers of ˙OH radicals (reaction (5)).107 For this reaction to be catalytic, the recycling of Fe(III) to Fe(II) according to reaction (6) is the rate-limiting step in the presence of H2O2.107 The kinetics of reaction (6) can be enhanced by light (reaction (10)) and in the presence of quinones and carboxylate ligands.107,115 The following section summarizes the key findings from photo-Fenton studies on models for HULIS that include gallic acid, catechol and guaiacol.Quici and Litter202 used a 15 W UV lamp with a maximum output at 366 nm to quantify the kinetics of gallic acid photodegradation on adding Fe(III) to a gallic acid–H2O2 mixture at pH 3. They reported a value for kUV of 0.049 min−1 compared with 0.0022 min−1 in the absence of H2O2. In another study, Quici et al.203 showed that the electrophilic addition of photoproduced ˙OH species to the aromatic ring in the presence of oxygen resulted in the release of CO2 and further oxidation of the gallic acid ring, which eventually resulted in ring opening and the formation of oxygenated organic compounds until the complete mineralization of gallic acid. Benitez et al.204 reported the photodegradation of gallic acid using a 15 W Hg lamp with a maximum output at 254 nm in the absence and presence of H2O2 and Fe(II) under neutral to acidic conditions. The highest kUV value (0.07 min−1) was reported when Fe(II) was added to an aqueous gallic acid–H2O2 mixture at pH 3.
Lofrano et al.205 investigated the conditions under which complete mineralization of catechol took place using Fenton reagents [H2O2 and Fe(II)] at pH 3 and irradiation with light. Using a 125 W fluorescent lamp with a maximum output at 350 nm and analysis by gas chromatography (GC) and UV-visible spectrophotometry, they found that 600:500 w/w of H2O2:Fe(II) and 30 min irradiation resulted in the significant removal of aromaticity, even when starting with relatively high catechol concentrations of up to 200 ppm. Another study reported that only oxalic acid was detected as a product by ion-exclusion HPLC from the photo-Fenton degradation of catechol, whereas a number of intermediates were identified by GC and ion-exclusion HPLC from the dark Fenton process. Using guaiacol, Samet et al.206 also reported the fastest degradation rates from photo-Fenton processes using the sun as the light source and 40% less H2O2 compared with the dark Fenton reaction. They noted that the photooxidation process initiated by ˙OH radicals formed a mixture of o- and p-quinone intermediates that absorbed light in the visible region. Further attack by ˙OH radicals resulted in ring opening and the formation of dicarboxylic acids and eventually CO2 and water.
In summary, the photochemical generation of ROS from hydroxylated iron species is pH dependent and efficient in transforming WSOC to variable degrees depending on their structure and the relative concentration of the oxidants. In the presence of light, the cycling between Fe(II) and Fe(III) becomes catalytic in nature to a certain point in the process. This photoreactivity could be complicated in the presence of other iron species with different ligands such as halogens, discussed in the following section.
5.2. Photochlorination of WSOC in the presence of iron and the production of halogens
The absorption of light by solvated Fe(III) in the presence of halogens such as Cl− ions results in the formation of chlorine radicals according to reaction (12):
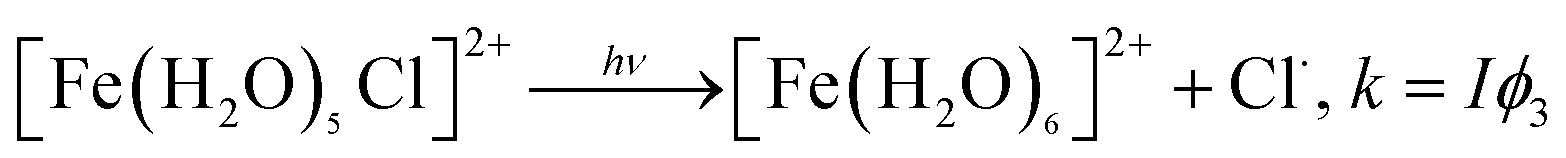 | (12) |
The quantum yields for reaction (12) vary depending on the experimental method. Lim et al.99 listed values between 0.093 and 0.13 from steady-state techniques and 0.46–0.57 from laser kinetic spectroscopy measurements. In addition, the photochemistry of hydroxylated Fe(III) species in the presence of Cl− ions yields Cl/Cl2˙− and ClOH˙− radicals, which, on formation, can undergo a number of bimolecular reactions with inorganic and organic species in solution. Table 1 lists the rate constants for reactions involving Cl and ˙OH radical species, adapted from Wittmer et al.101 The scavenging of ˙OH radicals by Cl− species competes with the formation of H2O2, explaining the overall inhibition of photo-Fenton reaction at high chloride concentrations (about 10 mM) under acidic conditions.207,208 The aforementioned Cl radicals are known as oxidants capable of electron transfer, hydrogen abstraction and additional reactions with organic compounds.209–213 For example, the bulk photochlorination of phenol210 and organic matter in forest soils was shown to proceed through these pathways and result in the formation of volatile organochlorine compounds.210,214 Keppler et al.215 reported the formation of C1–C4 organochlorine compounds in their studies on catechol degradation in the presence of Fe(III) and Cl−. The photoproduction of Cl2 gas from Fe(III) and Cl− solutions was also reported by Lim et al.99 using UV light at 365 nm.
| Reaction | kf (forward)/M−1 s−1 | kr (reverse)/s−1 |
|---|---|---|
OH˙ + Cl− ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) ClOH˙− ClOH˙− |
4.2 × 109 | 6.0 × 109 |
| ClOH˙− + H+ Cl˙ + H2O |
2.4 × 1010 | 1.8 × 105 |
| Cl˙ + Cl− Cl˙− |
7.8 × 109 | 5.7 × 104 |
| Cl˙ + Cl2˙− Cl− + Cl2 |
1.4 × 109 | |
| Cl2˙− + Cl2˙− → Cl3− + Cl− | 3.1 × 109 | |
| Cl3− Cl2 + Cl− |
K = 1.8 × 10−1 M−1 | |
| Cl˙ + Fe(II) → Cl− + Fe(III) | 5.9 × 109 | |
| Cl2˙− + Fe(II) → 2Cl− + Fe(III) | 1.4 × 107 | |
| ClOH˙− + Fe(II) → Cl− + OH− + Fe(III) | 1.3 × 108 | |
| OH˙ + Fe(II) → OH− + Fe(III) | 2.7 × 108 | |
| OH˙ + OH˙ → H2O2 | 5.2 × 109 |
Homogeneous and heterogeneous pathways that affect the formation of Cl2 gas are of great importance in atmospheric chemistry.216 Field studies have measured molecular halogens in coastal urban air at parts per trillion levels, which were mainly explained by known gas phase chemistry.217,218 Organic matter such as chlorophyll and aromatic ketones were shown to promote the heterogeneous photooxidation of halide ions at the air–salt water interface.219,220 In a related study, Ofner et al.221 showed that reactive chlorine and bromine species react with SOA particles derived from α-pinene, catechol and guaiacol, which, in turn, changed their chemical (i.e. structure) and physical (i.e. size and optical) properties. Recently, Wittmer et al.101 reported an order of magnitude increase in Cl2 gas release from an Fe(III)-doped NaCl salt pan relative to plain NaCl samples on irradiation. The same group also observed the inhibition in Cl2 gas release from the former samples on adding sulfate, oxalate and catechol, which was explained by the strong complexation of these ligands with Fe(III). An inhibition in the release of brominated and chlorinated gaseous species was observed in the presence of the SOA precursors catechol and guaiacol (no iron) on irradiation of a simulated salt pan.221 To summarize, in multicomponent atmospheric aerosols containing Fe(III), Cl− and organics, the aforementioned bulk chemistry influences the photooxidation of halide ions and the nature of the organic phase. Although bulk phase chemistry is relevant for the micron-size droplets common in clouds, surface contributions become more important as the particle size decreases and, as detailed in the following section, are explained by different mechanisms from the bulk processes.
6. Surface photochemistry of iron
Current cloud chemistry models contain thermodynamic and kinetic parameters from known photochemical reactions in the bulk phase.222 These reactions do not necessarily and accurately represent photochemical reactions occurring at the surface of aerosols or on atmospherically relevant surfaces.17 Questions still remain about the relative efficiency of interfacial Fe(III) photochemistry in degrading organic matter in the presence of a few layers of adsorbed water compared with that in bulk liquid water under photon fluxes that simulate the solar flux. Answering these questions demands molecular level information on the nature of the surface water (see Section 4.1) and the photochemistry of multicomponent surfaces containing transition metals such as Fe(III) in their soluble and insoluble forms. The following sections highlight recent studies in these areas.6.1. Solvent cage effect and surface iron
In small droplets or at the interface of systems containing chromophores such as iron, an observed overall increase in quantum yield is expected for radical formation.223 This increase is explained by the enhancement in the surface concentration of reactants, increases in the light intensity due to morphology dependent resonances and/or refraction, and/or decreased solvent cage effects.223 Nissenson et al.192,223 demonstrated through experiments and modelling that the third factor is the major contributing factor after accounting for the surface enrichment of the organic species and the distribution of light throughout the droplet. One experiment was the photolysis of molybdenum hexacarbonyl, Mo(CO)6, in 1-decene, either as liquid droplets or in bulk liquid solutions, where the results showed that the rates in the aerosols were faster by at least three orders of magnitude than in the bulk liquids.223 The modelling study was carried out on cloud droplets containing benzene and [Fe(OH)]2+ as a photosensitizer that produced ˙OH radicals on irradiation.192 The average angle and wavelength-relative light intensities were calculated as a function of distance from the droplet centre for three droplet radii of 1–3 μm. The results showed that the calculated light intensity decreased at the surface. The overall rate equation for the decay of benzene as a result of this chemistry included the rate equations for all possible reactions, such as reactions with scavengers in cloud droplets. The distribution of the reaction rate within the droplet volume was calculated from the integration of this equation over the radius range. The results of this exercise showed a sharp increase in the decay rate of benzene at the interface, where the fraction of the total reaction in the surface layer was calculated to be 35 and 15% for droplets with radii of 1 and 3 μm, respectively. After accounting for the surface enhancement of benzene and the distribution of light throughout the droplet, the increase in the surface reaction rate was explained by the increase in the photolysis quantum yield due to the decreased solvent cage.Some research groups in North America and Europe have studied the efficiency of surface-photosensitized reactions in metal-free organic-containing aerosols. This photochemistry can lead to the oxidation of surface organics, an enhancement in uptake and the production of gas phase species, and the formation of SOA.19 Examples include (but are not limited to) the following: Karagulian et al.224 investigated the “bottom-up” photooxidation of a fatty acid adsorbed on NaCl mixed with solid NO2− and reported the formation of organic products consistent with either an O− or ˙OH radical attack depending on the amount of water. Cowen and Al-Abadleh36 proposed mechanism for the formation of surface aryl aldehydes by irradiating solid tannic acid under humid conditions using DRIFTS. Kleffmann and co-workers measured the fast photosensitized formation of HONO from the reaction of NO2 on humic acids43 and mineral dust225 under humid conditions. Donaldson and co-workers reported the photoenhanced uptake of NO2 and O3 and the oxidation of halides by chlorophyll at the salt water–air interface.219,226,227 In addition, a new photoinduced pathway for particle growth was reported by Monge et al.228 as a result of the photoenhanced uptake of terpenes (limonene and isoprene) on seed particles containing humic acid, succinic acid and ammonium nitrate (1:10:1 in weight). In these experiments, humic acid acted as the main photosensitizer and the formation of the triplet state produced radicals at the surface of aerosols, which enhanced the uptake of non-condensable terpenes.
Very little experimental work has explored the extent to which the known Fenton and photo-Fenton chemistry takes place at the gas–solid interface in the presence of organic matter and a few layers of adsorbed water. The following section highlights related studies that aimed to fill this knowledge gap.
6.2. Heterogeneous photochemistry of insoluble iron in the presence of organic species
Iron (oxyhydr)oxides have band gaps in the visible region of the electromagnetic spectrum. For example, the band gap for hematite is around 2.2 eV (565 nm)229 and that of goethite ranges from 2.1 to 2.5 eV (592–497 nm);166 these band gaps have been found to increase with decreasing particle size.230,231 Therefore band gap excitation using UV photons with energies >2.1 eV leads to the creation of electron–hole pairs followed by the formation of Fe(II) species. The photoreactivity of iron (oxyhydr)oxides towards inorganic gases such as nitrogen and sulfur oxides have been shown to be important for the renoxification and oxidation of sulfur dioxide.232 The absorption of light by adsorbed organic species initiates LMCT to surfaceFe(III), leading to the formation of reduced iron sites, Fe(II), which then desorb to the bulk phase (i.e. photoreductive dissolution). The formation of electron–hole pairs in the presence of organic species can also initiate redox chemistry, leading to the transformation of these materials.13
The photoreductive dissolution of iron (oxyhydr)oxides in natural water and seawater has received much attention and was reviewed by Zwiener and Frimmel233 and Baker and Croot.234 Similar studies were conducted with iron-containing coal fly ash and Arizona test dust as representatives of anthropogenic and natural mineral dusts.181 Briefly, the studies showed the direct photoreductive formation of Fe(II) or organic-iron complexes in photoreductive experiments, with the highest rates observed at low pH values. Fe(II) was subsequently oxidized to Fe(III), which reforms smaller iron colloids or binds to organic chelators. The presence of organic ligands enhances this process, particularly at low pH values. The indirect reduction (and oxidation) of iron species also takes place as a result of reactions with ROS such as superoxide (O2−) and hydrogen peroxide (H2O2) produced from photochemical reactions involving dissolved organic matter in seawater and enzymatic action in bacteria and phytoplankton. The cycling between Fe(II) and Fe(III), as summarized earlier, explains the apparent extended lifetime of Fe(II) in iron enrichment experiments in the Southern Ocean.
Some reports have shown that the photoreductive dissolution of iron (oxyhydr)oxides was accelerated for particles trapped in ice. Kim et al.185 measured the concentration of dissolved Fe(II) from the dissolution of hematite, goethite and maghemite in ice samples under UV and visible irradiation in the presence and absence of organic electron donors under acidic pH conditions and compared the results with measurements from dark experiments and similar experiments in bulk liquid water. The results showed an enhancement in the photogeneration of dissolved Fe(II) in ice regardless of the type of iron material or the organic ligand, with higher rates using UV rather than visible light. This is in contrast with dark dissolution in ice, where Fe(III) was mainly formed and its formation rate depended on the type of organic ligand.184 The photochemical mechanism was explained with the following equations for the case of hematite, which can be generalized for other iron (oxyhydr)oxides:185
|
Fe(III)–L + hν → Fe(II)–L˙+ (photoinduced LMCT)
| (13) |
| Fe2O3 + hν → ecb− + hvb+ (bandgap excitation) | (14) |
| Fe(III) (at lattice or surface site) + ecb− → Fe(II)surf | (15) |
| Fe(II) (at lattice or surface site) → Fe(II)aq | (16) |
The photocatalytic reactivity of iron (oxyhydr)oxides and other metal oxides was demonstrated in a number of studies that involved gas phase and dissolved species of importance to atmospheric chemistry.12–14,195,235 Examples included the photoenhanced uptake of NO2 and the formation of HONO on mineral dust,225 the photoenhanced uptake of O3 by dust,236 the photoenhanced uptake of formaldehyde237,238 and short-chain alcohols239 on TiO2, Fe2O3, dust and volcanic ash. The mechanisms that explain these observations involve the generation of electron–hole pairs as a result of the absorption of light, which serves as the driver for charge transfer reactions with adsorbed water, NO2 and O3. The main radical that forms from the reaction of adsorbed water with the electron–hole pairs generated on irradiation is the ˙OH radical, which seems to contribute to the photoenhanced uptake of VOCs. This photouptake was reported to be dependent on the RH, as in the case of formaldehyde,238 where the uptake coefficient was at a maximum at 30% RH and then decreased at higher values as a result of site blockage by adsorbed water. Nitrite and chloride anions are also reduced by reaction with holes, which, in the presence of aromatic compounds, can lead to nitration and chlorination of the benzene ring.13 The following section shows the differences between these studies and those involving soluble iron.
6.3. Heterogeneous photochemistry of soluble iron in the presence of organic compounds
The mechanisms that explain the reactivity of coordinated Fe(III) in iron (oxyhydr)oxides do not apply to solvated Fe(III) cations in hygroscopic iron salts. In a set of experiments probing the gas–aqueous interface, Rifkha Kameel et al.240 reported the Fenton oxidation of gaseous isoprene using acidic FeCl2 aqueous microjets. In these experiments, the gaseous reactant streams, isoprene and H2O2 intersected the droplets containing iron for about 10 μs and the products were analysed in situ via online electrospray ionization mass spectrometry. The results were consistent with an oxidation process initiated by the addition of ˙OH radicals to protonated isoprene oligomer homologues, followed by fast reactions involving dissolved H2O2, HO2˙ and O2 that led to the formation of polyols, carbonyls and, to a lesser extent, carboxylic acids in the condensed phase. When taking into account typical values for the uptake coefficients of the reactants, the concentrations and Henry's law constants, it was concluded that this iron-driven pathway for the oxidation of VOCs and the formation of SOA was potentially important during both the day and at night.Tofan-Lazar and Al-Abadleh30 examined the role played by UV-visible light in the uptake of gas phase catechol by samples containing FeCl3 particles. Experiments were conducted such that the solid sample was irradiated before the introduction of catechol vapour under humid or dry conditions; this was followed by the introduction of catechol vapour under continuous irradiation. The solid lines in Fig. 20a show the spectra of surface catechol as a function of the irradiation time at 30% RH, whereas the dashed lines represent the spectra collected under dry conditions (<1% RH). These spectra are different from those shown for the uptake of catechol under dark conditions (humid and dry, Fig. 20b). Overlapping spectral features between 1600 and 1000 cm−1 were assigned previously31 as evidence for the formation of catechol–Fe complexes under humid conditions based on comparison with the infrared spectra of these compounds in bulk solution recorded as a function of the pH value. The solid lines in Fig. 20a represent the net of two processes: the continuous uptake of catechol from the gas phase and photodegradation. The kinetic curves shown in Fig. 20c were best described using an empirical sigmoidal growth equation (see supporting information in Tofan-Lazar and Al-Abadleh30 for equation, best-fit parameters and the calculated growth factors). Within the uncertainty of these measurements, the growth factors calculated at 30% RH under light and dark conditions were very similar and were higher by nearly a factor of three than in dry conditions over a period of 60 min (light and dark). This suggests that light does not significantly enhance the uptake of catechol vapour on FeCl3 particles and that surface water in equilibrium with 30% RH (in the light and dark) increases the surface concentration of catechol.
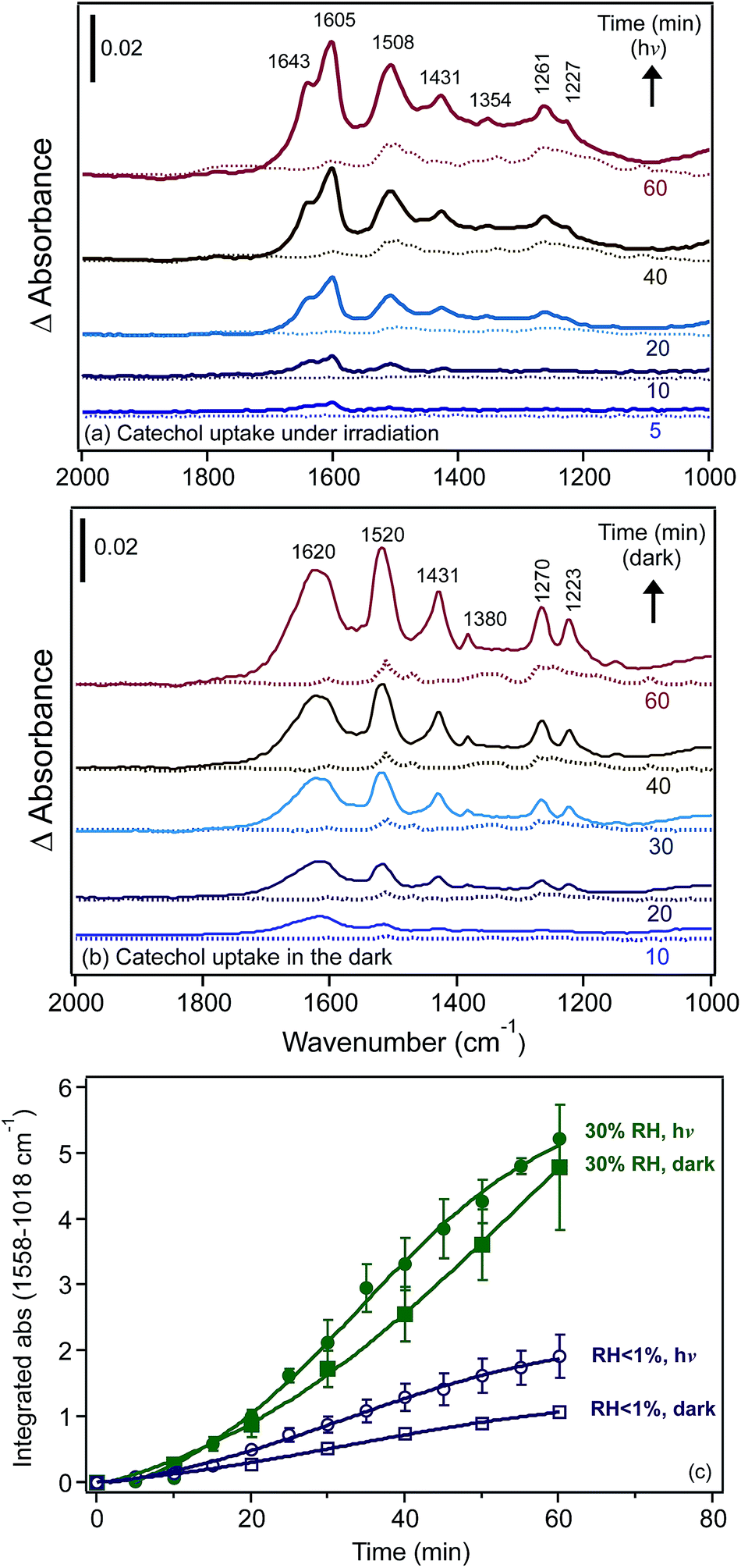 | ||
| Fig. 20 Representative time-dependent DRIFTS absorbance spectra of surface catechol on 1% FeCl3(s) as a result of flowing gas phase catechol in the dark under (a) irradiation and (b) in the dark. Solid and dashed lines show the spectra collected under humid (at 30% RH) and dry conditions (RH < 1%), respectively. The gas phase concentration of catechol is estimated as 32 ppb. (c) Kinetic curves showing the increase in the integrated absorbance from 1558 to 1018 cm−1 assigned to surface catechol in the presence of 1% FeCl3(s) as a result of flowing gas phase catechol under dry and humid conditions, both in the dark and irradiation. Error bars represent ±σ from averaging two experiments. abs = absorbance in y-axis label. Reprinted with permission from ref. 30 (Copyright © 2014, American Chemical Society). | ||
The irradiation of solid FeCl3 samples containing adsorbed catechol under a constant flow of humid or dry air only (i.e. no gas phase catechol) gave difference infrared spectra that showed the breakdown of the surface catechol–Fe complexes with increasing irradiation time (Fig. 21a).30 This photodecay can be explained by eqn (10)–(12) because the enhanced ionic mobility in the presence of surface water at 30% RH explained the presence of the reactant Fe species. We believe that reaction (12), producing ˙Cl radicals, is responsible for the decay of the catechol–Fe complexes for the following reasons: (a) the high acidity of the “quasi liquid” phase at 30% RH as a result of FeCl3 dissolution drives a higher concentration of [Fe(H2O)5Cl]2+ (about 1 M, pH 1) relative to [Fe(H2O)5(OH)]2+ (about 0.1 M, pH 1), as calculated from the bulk solubility of FeCl3 (92 g/100 mL in water at 25 °C)241 and the bulk aqueous phase speciation curve shown in Fig. 4b the higher quantum yields for the photodissociation of [Fe(H2O)5Cl]2+ versus [Fe(H2O)5(OH)]2+ in the bulk aqueous phase (i.e. with a solvent cage) (see Section 6.1) – these values are predicted to be higher at the gas–solid interface in the presence of adsorbed water; and (c) given the relatively low gas phase concentration of catechol in these experiments (estimated at 32 ppb (ref. 31)) and an equilibrium constant for catechol–Fe complex formation of 4.32 × 10−2 M,242 the concentration of [Fe–cat]2+ is around 4 mM, assuming a pH of 1. In addition, catechol–Fe complexes are more susceptible to the electrophilic additions of ˙Cl radicals to the aromatic ring than uncomplexed catechol as a result of LMCT in the former. The stretching absorptions of C–Cl bonds that indicate the chlorination of catechol would occur below 800 cm−1. These absorptions were not observed in our data because they were obstructed by the liberational modes of surface water. These modes were most intense below 1000 cm−1 for bulk liquid water243 and ice244 and are known to blue shift and increase in intensity and bandwidth with increasing strength of hydrogen bonding.245 The absence of carbonyl stretching modes between 1800 and 1700 cm−1 in Fig. 21a also suggested that there was no formation of quinone, ketone or aldehyde species, confirming negligible oxidation by photogenerated ˙OH radicals.
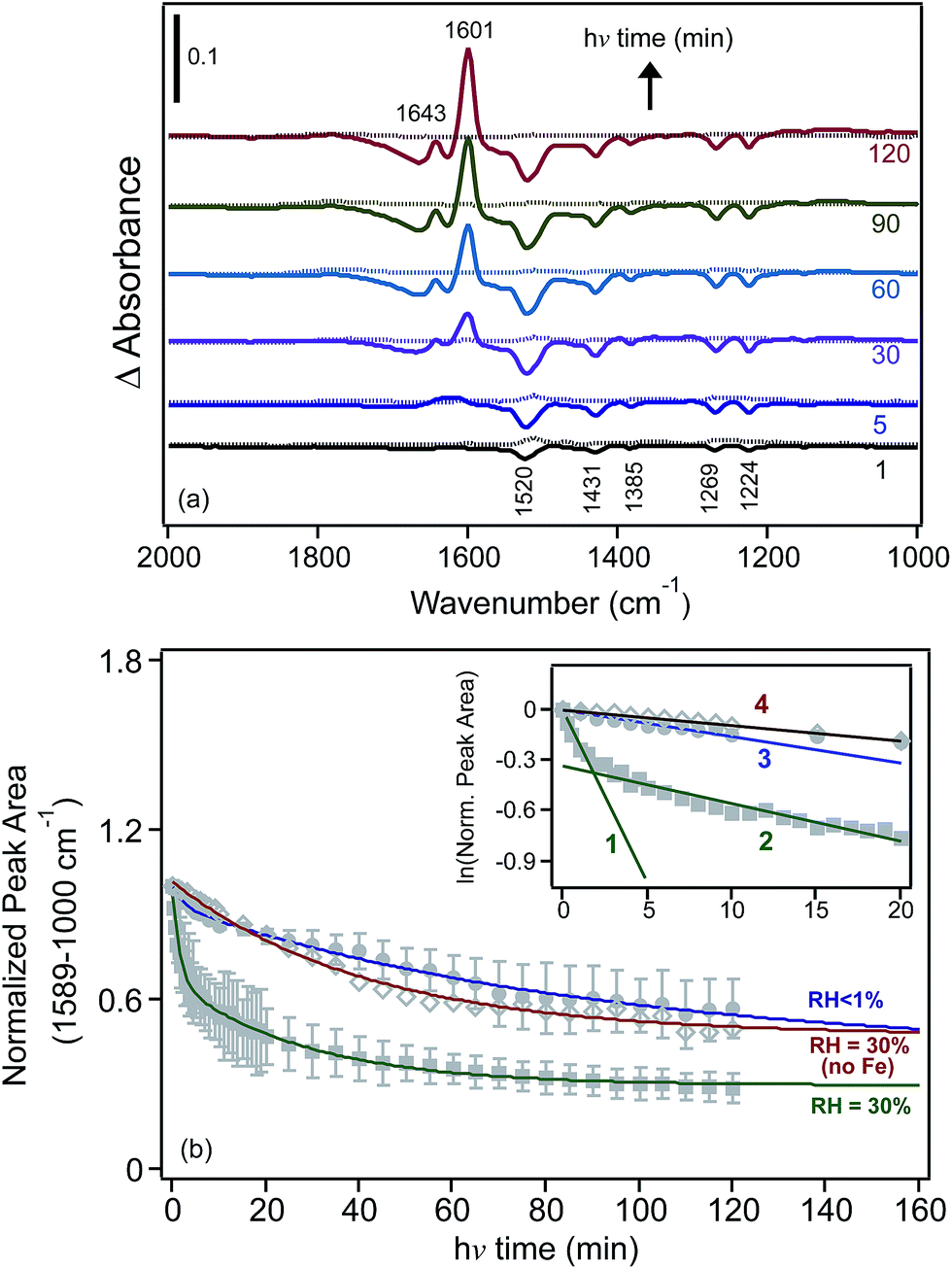 | ||
| Fig. 21 (a) Difference DRIFTS absorbance spectra showing the photoreactivity of surface catechol in the presence of 1% FeCl3(s) as a result of irradiation under humid (at 30% RH, solid lines) and dry (at RH < 1%, dashed lines) conditions. The FeCl3 samples were first exposed to gas phase catechol in the dark for 60 min, then the catechol flow was turned off while maintaining a humid or dry air flow over the sample during the irradiation part of the experiment. (b) Kinetic curves showing the decrease in the normalized integrated absorbance from 1589 to 1000 cm−1 assigned to surface catechol in the absence and presence of 1% FeCl3(s) as a result of irradiation under humid and dry conditions. Solid markers represent experimental data collected under dry and humid conditions. Open markers represent data from a control experiment with no Fe in the sample. Lines represent least-squares fittings using double-exponential equations. The inset shows experimental data plotted in the linearized first-order kinetics form for the first 20 min of irradiation. The values of slopes represent the apparent photodecay constant (min−1): 0.21 ± 0.03 (line 1), 0.023 ± 0.003 (line 2), 0.016 ± 0.002 (line 3) and 0.009 ± 0.001 (line 4). Error bars represent ±σ from the averaging of two experiments. Reprinted with permission from ref. 30 (Copyright © 2014, American Chemical Society). | ||
Fig. 21b shows the normalized kinetic curves under humid conditions and control experiments for the photodecay of adsorbed catechol under dry conditions (RH < 1%) and with no FeCl3 at 30% RH. The inset shows the natural logarithm of these data and the best-fit slopes corresponding to the apparent photodecay constants as listed in the caption. This analysis clearly showed that surface water enhances the initial photodecay kinetics of catechol–Fe complexes at the solid–humid air interface by a factor of ten relative to control experiments with no FeCl3 under humid conditions or with dry FeCl3. The slower rate observed under humid conditions at longer irradiation times suggested a contribution from another reaction that caused the photodecay of the catechol–Fe complexes. Hence the main two ingredients for the fast photodecay of organic compounds to take place were solvated Fe(III) and surface water in equilibrium with at least 30% RH. The lifetime of the organic compounds was estimated to be 38 min as a result of this heterogeneous pathway, as calculated in the supporting information of Tofan-Lazar and Al-Abadleh,30 which is about 1000 times shorter than that calculated for the fastest homogeneous reaction of ˙Cl radicals with organic compounds.
In summary, as observed in the dark, this section has illustrated the key mechanistic differences in the surface photochemical reactivity of iron towards the transformation of organic matter, which depends on the source of the iron (hygroscopic versus insoluble material) and the amount of surface water.
7. Summary and future directions
This review has synthesized the current state of knowledge of iron chemistry in multicomponent atmospheric aerosols, particularly those containing HULIS and their model compounds. Field measurements have focused on the sources, characterization and fate of iron to the oceans and have led to the improvement of a number of models. Laboratory studies have highlighted the complexity of iron reactivity, which varies depending on the solubility, the redox conditions, the absence and presence of UV-visible light, the presence of organic species and ROS, the pH and temperature. Several examples have shown key differences between the bulk and surface chemistry of iron-containing materials, which varied considerably as a result of the solvent cage effect and the structure of water.Elucidation of the mechanisms and the extraction of kinetic and thermodynamic parameters are necessary for photochemical reactions occurring at the surface and in the bulk of multicomponent aerosol systems containing transition metals, such as iron, and organic matter. These experiments need to be conducted under the atmospherically relevant conditions of temperature, RH, the wavelength and intensity of light, and over relatively short and long timeframes, taking into account the phase, size, water content, oxidant content and acidity of model aerosol systems. These studies need also to be complemented with analytical techniques or procedures capable of identifying and quantifying the gas phase species that are either consumed or released. Working with the different classes of organic functional groups identified in ambient aerosols will provide correlations with the rates and mechanisms that are hard to extract when working with large multifunctional macromolecules. Hence, when characterizing this latter class of molecule, either as model compounds or collected from field campaigns, it is insightful to report the amount and type of organic functional groups in addition to the O:C ratios. Comparison of the results obtained from photochemistry with those obtained in dark conditions will complete the picture on the relative importance of reactions during the day and at night.
Insights from computational chemistry on iron–water clusters in the presence of other inorganic and organic solutes would be invaluable in understanding the interfacial regions of these systems. These results would need to be coupled with the more accurate quantification and characterization of soluble iron and organic matter in field samples. Such an integrated approach would improve the predictive power of climate and cloud chemistry models with respect to the heterogeneous dark and photochemistry of aerosols and other atmospherically important iron-containing surfaces and their role in generating SOA, altering the balance of gas phase species and increasing the solubility of iron in different materials.
Acknowledgements
The author acknowledges contributions to experiments and data analysis from the following students at Laurier: Scott Cowen, Gregory Wentworth, Julia Tofan-Lazar, Arthur Situm, Samantha Slikboer, Lindsay Grandy and Kevin Jakiela. The author is also grateful for stimulating discussions and fruitful collaborations with Professor Sergey Nizkorodov and Sandra Blair (University of California, Irvine), Dr Richard Smith (University of Waterloo), Professor James Donaldson (University of Toronto) and Professor Cornelius Zetzsch (University of Bayreuth). Financial support to the atmospheric chemistry research program at Laurier was provided by Wilfrid Laurier University, the Natural Science and Research Council of Canada (NSERC) Discovery and Research Tools and Instruments programs, the Canadian Foundation for Innovation Leaders Opportunities Fund (CFI-LOF) and an Ontario Ministry of Research and Innovation Early Researcher Award (MRI-ERA).References
- B. J. Finlayson-Pitts and J. N. Pitts Jr, Chemistry of the Upper and Lower Atmosphere, Academic Press, New York, 2000 Search PubMed.
- B. A. Maher, J. M. Prospero, D. Mackie, D. Gaiero, P. P. Hesse and Y. Balkanski, Earth-Sci. Rev., 2010, 99, 61–97 CrossRef CAS PubMed.
- N. Mahowald, Science, 2011, 334, 794–796 CrossRef CAS PubMed.
- P. Ciesla, P. Kocot, P. Mytych and Z. Stasicka, J. Mol. Catal. A: Chem., 2004, 224, 17–33 CrossRef CAS PubMed.
- A. Krishnamurthy, J. K. Moore, N. Mahowald, C. Luo and C. S. Zender, J. Geophys. Res., 2010, 115, G01006, DOI:10.1029/2009jg001115.
- IPCC, Summary for Policymakers, in Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change Stocker, ed. T. F. Stocker, D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex and P. M. Midgley, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2013 Search PubMed.
- S.-S. Lee, Nat. Geosci., 2011, 4, 826–827 CrossRef CAS PubMed.
- S. R. Suda, M. D. Petters, G. K. Yeh, C. Strollo, A. Matsunaga, A. Faulhaber, P. J. Ziemann, A. J. Prenni, C. M. Carrico, R. C. Sullivan and S. M. Kreidenweis, Environ. Sci. Technol., 2014, 48, 10182–10190 CrossRef CAS PubMed.
- J. M. Sun and P. A. Ariya, Atmos. Environ., 2006, 40, 795–820 CrossRef CAS PubMed.
- R. Zhang, A. Khalizov, L. Wang, M. Hu and W. Xu, Chem. Rev., 2012, 112, 1957–2011 CrossRef CAS PubMed.
- P. J. Ziemann and R. Atkinson, Chem. Soc. Rev., 2012, 41, 6582–6605 RSC.
- J. Ma, Y. Liu, C. Han, Q. Ma, C. Liu and H. He, J. Environ. Sci., 2013, 25, 326–334 CrossRef CAS.
- D. Vione, V. Maurino, C. Minero, E. Pelizzetti, M. A. J. Harrison, R.-I. Olariu and C. Arsene, Chem. Soc. Rev., 2006, 35, 441–453 CAS.
- D. M. Cwiertny, M. A. Young and V. H. Grassian, Annu. Rev. Phys. Chem., 2008, 59, 27–51 CrossRef CAS PubMed.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, Wiley, New York, 2006 Search PubMed.
- M. R. Hoffmann, Homogeneous and Heterogeneous Photochemistry in the Troposphere, in Environmental Photochemistry Part II, ed. P. Boule, D. W. Bahnemann and P. Robertson, Springer, Berlin, 2005, vol. 2M, pp. 77–118 Search PubMed.
- B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2009, 11, 7760–7779 RSC.
- E. G. Alvarez, H. Wortham, R. S. Strekowski, C. Zetzsch and S. Gligorovski, Environ. Sci. Technol., 2012, 46, 1955–1963 CrossRef PubMed.
- C. George, M. Ammann, B. D'Anna, D. J. Donaldson and S. A. Nizkorodov, Chem. Rev., 2015 DOI:10.1021/cr500648z.
- E. R. Graber and Y. Rudich, Atmos. Chem. Phys., 2006, 6, 729–753 CrossRef CAS.
- G. Zheng, K. He, F. Duan, Y. Cheng and Y. Ma, Environ. Pollut., 2013, 181, 301–314 CrossRef CAS PubMed.
- L. I. Nieto-Gligorovski, S. Net, S. Gligorovski, H. Wortham, H. Grothe and C. Zetzsch, Atmos. Environ., 2010, 44, 5451–5459 CrossRef CAS PubMed.
- J. Ofner, H.-U. Kruger, H. Grothe, P. Schmitt-Kopplin, K. Whitmore and C. Zetzsch, Atmos. Chem. Phys., 2011, 11, 1–15 CrossRef CAS PubMed.
- R. M. B. O. Duarte, E. B. H. Santos, C. A. Pio and A. C. Duarte, Atmos. Environ., 2007, 41, 8100–8113 CrossRef CAS PubMed.
- K. M. Prather, C. D. Hatch and V. H. Grassian, Annu. Rev. Anal. Chem., 2008, 1, 485–514 CrossRef CAS PubMed.
- V. Zelenya, T. Huthwelker, A. Krepelova, Y. Rudich and M. Ammann, Environ. Chem., 2011, 8, 450–460 Search PubMed.
- S. Cowen, Heterogeneous Photodegradation of Tannic Acid Studied by Diffuse Reflectance FTIR, MSc thesis, University of Guelph, 2009 Search PubMed.
- Y. Sosedova, A. Rouviere, T. Bartels-Rausch and M. Ammann, Photochem. Photobiol. Sci., 2011, 10, 1680–1690 CAS.
- G. Wentworth and H. A. Al-Abadleh, Phys. Chem. Chem. Phys., 2011, 13, 6507–6516 RSC.
- J. Tofan-Lazar and H. A. Al-Abadleh, Environ. Sci. Technol., 2014, 48, 394–402 CrossRef CAS PubMed.
- J. Tofan-Lazar, A. Situm and H. A. Al-Abadleh, J. Phys. Chem. A, 2013, 117, 10368–10380 CrossRef CAS PubMed.
- Z. Kitanovski, A. Cusak, I. Grgic and M. Claeys, Atmos. Meas. Tech., 2014, 7, 2457–2470 CAS.
- S. S. Steimer, M. Lampimaki, E. Coz, G. Grzinic and M. Ammann, Atmos. Chem. Phys., 2014, 14, 10761–10772 Search PubMed.
- M. N. Chan and C. K. Chan, Environ. Sci. Technol., 2003, 37, 5109–5155 CrossRef CAS.
- C. D. Hatch, K. M. Gierlus, J. D. Schuttlefield and V. H. Grassian, Atmos. Environ., 2008, 42, 5672–5684 CrossRef CAS PubMed.
- S. Cowen and H. A. Al-Abadleh, Phys. Chem. Chem. Phys., 2009, 11, 7838–7847 RSC.
- E. Dinar, I. Taraniuk, E. R. Graber, T. Anttila, T. F. Mentel and Y. Rudich, J. Geophys. Res.: Atmos., 2007, 112, D05211 CrossRef.
- M. N. Chan, S. M. Kreidenweis and C. K. Chan, Environ. Sci. Technol., 2008, 42, 3602–3608 CrossRef CAS.
- C. Richard and S. Canonica, Aquatic phototransformation of organic contaminants induced by coloured dissolved organic matter, in Hdb Environmental Chemistry, Springer-Verlag, Berlin, 2005, vol. 2, pp. 299–323 Search PubMed.
- M. A. Young, Environmental Photochemistry in Surface Waters, in Water Encyclopedia: Oceanography, Meteorology, Physics and Chemistry, Water Law, and Water History, Art, and Culture, ed. J. Lehr and J. Kelley, Wiley & Sons, New York, 2005, pp. 529–540 Search PubMed.
- M. C. Gonzalez and E. S. Roman, Environmental photochemistry in heterogeneous media, in Hdb Environmental Chemistry, Springer-Verlag, Berlin, 2005, vol. 2, pp. 49–75 Search PubMed.
- K. Stemmler, M. Ammann, C. Donders, J. Kleffmann and C. George, Nature, 2006, 440, 195–198 CrossRef CAS PubMed.
- J. Kleffmann, ChemPhysChem, 2007, 8, 1137–1144 CrossRef CAS PubMed.
- M. Shiraiwa, M. Ammann, T. Koop and U. Pöschl, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11003–11008 CrossRef CAS PubMed.
- M. Kuwata and S. T. Martin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17354–17359 CrossRef CAS PubMed.
- Y. You, L. Renbaum-Wolff, M. C. Sospedra, S. J. Hanna, N. Hiranuma, S. Kamal, M. L. Smith, X. Zhang, R. J. Weber and J. E. Shilling, et al., Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13188–13193 CrossRef CAS PubMed.
- Y. Rudich, N. M. Donahue and T. Mentel, Annu. Rev. Phys. Chem., 2007, 58, 321–352 CrossRef CAS PubMed.
- A. L. Robinson, N. M. Donahue, M. K. Shrivastava, E. A. Weitkamp, A. M. Sage, A. P. Grieshop, T. E. Lane, J. R. Pierce and S. N. Pandis, Science, 2007, 315, 1259–1262 CrossRef CAS PubMed.
- J. H. Kroll and J. H. Seinfeld, Atmos. Environ., 2008, 42, 3593–3624 CrossRef CAS PubMed.
- R. Volkamer, P. J. Ziemann and M. J. Molina, Atmos. Chem. Phys., 2009, 9, 1907–1928 CrossRef CAS.
- I. J. George, J. Slowik and J. P. D. Abbatt, Geophys. Res. Lett., 2008, 35, L13811 CrossRef.
- E. Harris, B. Sinha, D. van Pinxteren, A. Tilgner, K. W. Fomba, J. Schneider, A. Roth, T. Gnauk, B. Fahlbusch, S. Mertes, T. Lee, J. Collett, S. Foley, S. Borrmann, P. Hoppe and H. Herrmann, Science, 2013, 340, 727–730 CrossRef CAS PubMed.
- J. G. Charrier and C. Anastasio, Atmos. Chem. Phys., 2012, 12, 9321–9333 CAS.
- D. A. S. Finden, E. Tipping, G. H. M. Jaworski and C. S. Reynolds, Nature, 1984, 309, 783–784 CrossRef CAS PubMed.
- T. D. Waite and F. M. M. Morel, Environ. Sci. Technol., 1984, 18, 860–868 CrossRef CAS PubMed.
- N. Mahowald, A. R. Baker, G. Bergametti, N. Brooks, R. A. Duce, T. D. Jickells, N. Kubilay, J. M. Prospero and I. Tegen, Global Biogeochem. Cycles, 2005, 19, GB4025 Search PubMed.
- T. M. Conway and S. G. John, Nature, 2014, 511, 212–215 CrossRef CAS PubMed.
- P. N. Sedwick, E. r. Sholkovitz and T. M. Church, Geochem., Geophys., Geosyst., 2007, 8, Q10Q06, DOI:10.1029/2007gc001586.
- C. Guieu, S. Bonnet, T. Wagener and M.-D. Loye-Pilot, Geophys. Res. Lett., 2005, 32, L19608, DOI:10.1029/2005gl022962.
- A. Ito, Environ. Sci. Technol. Lett., 2015, 2, 70–75 CrossRef CAS.
- R. Benner, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 893–894 CrossRef CAS PubMed.
- P. W. Boyd and M. J. Ellwood, Nat. Geosci., 2010, 3, 675–682 CrossRef CAS PubMed.
- A. W. Schroth, J. Crusius, E. R. Sholkovitz and B. C. Bostick, Nat. Geosci., 2009, 2, 337–340 CrossRef CAS PubMed.
- J. H. Martin, R. M. Gordon and S. E. Fitzwater, Limnol. Oceanogr., 1991, 36, 1793–1802 CrossRef.
- J. K. Moore, S. C. Doney, D. M. Glover and I. Y. Fung, Deep Sea Res., Part I, 2002, 49, 463–507 CrossRef CAS.
- J. K. Moore and O. Braucher, Biogeosciences, 2008, 5, 631–656 CAS.
- C.-E. Thuroczy, L. J. A. Gerringa, M. Klunder, P. Laan, M. Le Guitton and J. W. de Baar, J. Geophys. Res., 2011, 116, C10009, DOI:10.1029/2010jc006835.
- B. P. von der Heyden, A. N. Roychoudhury, T. N. Mtshali, T. Tyliszczak and S. C. B. Myneni, Science, 2012, 338, 1199–1201 CrossRef CAS PubMed.
- M. Oakes, R. J. Weber, B. Lai, A. Russell and E. D. Ingall, Atmos. Chem. Phys., 2012, 12, 745–756 CAS.
- A. J. Butler, M. S. Andrew and A. G. Russell, J. Geophys. Res.: Atmos., 2003, 108, SOS 3-1–SOS 3–11 CrossRef.
- P. Hoffmann, A. N. Dedik, J. Ensling, S. Weinbruch, S. Weber, T. Sinner, P. Gutlich and H. M. Ortner, J. Aerosol Sci., 1996, 27, 325–327 CrossRef CAS.
- A. M. Johansen, R. Siefert and M. R. Hoffmann, J. Geophys. Res.: Atmos., 2000, 105, 15277–15312 CrossRef CAS.
- W. Liu, Y. H. Wang, A. Russell and E. S. Edgerton, Atmos. Environ., 2005, 39, 4453–4470 CrossRef CAS PubMed.
- M. Oakes, N. Rastogi, B. J. Majestic, M. Shafer, J. J. Schauer, E. S. Edgerton and R. J. Weber, J. Geophys. Res.: Atmos., 2010, 115, 1–12 CrossRef.
- A. P. Ault, T. M. Peters, E. J. Sawvel, G. S. Casuccio, R. D. Willis, G. A. Norris and V. H. Grassian, Environ. Sci. Technol., 2012, 46, 4331–4339 CrossRef CAS PubMed.
- T. L. Guasco, L. A. Cuadra-Rodriguez, B. E. Pedler, A. P. Ault, D. B. Collins, D. Zhao, M. J. Kim, M. J. Ruppel, S. C. Wilson, R. S. Pomeroy, V. H. Grassian, F. Azam, T. H. Bertram and K. A. Prather, Environ. Sci. Technol., 2014, 48, 1324–1333 CrossRef CAS PubMed.
- A. P. Ault, M. J. Moore, H. Furutani and K. A. Prather, Environ. Sci. Technol., 2009, 43, 3500–3506 CrossRef CAS.
- A. P. Ault, C. J. Gaston, Y. Wang, G. Dominguez, M. H. Thiemen and K. A. Prather, Environ. Sci. Technol., 2010, 44, 1954–1961 CrossRef CAS PubMed.
- K. Deboudt, P. Flament, M. Choel, A. Gloter, S. Sobanska and C. Colliex, J. Geophys. Res.: Atmos., 2010, 115, D24207 CrossRef.
- S. Takahama, S. Gilardoni and L. M. Russell, J. Geophys. Res., 2008, 113, D22202, DOI:10.1029/2008jd009810.
- A. P. Ault, T. L. Guasco, O. S. Ryder, J. Baltrusaitis, L. A. Cuadra-Rodriguez, D. B. Collin, M. J. Ruppel, T. H. Bertram, K. A. Prather and V. H. Grassian, J. Am. Chem. Soc., 2013, 135, 14528–14531 CrossRef CAS PubMed.
- B. Gantt and N. Meskhidze, Atmos. Chem. Phys., 2013, 13, 3979–3996 Search PubMed.
- C. Oppo, S. Bellandi, N. D. Innocenti, A. M. Stortini, G. Loglio, E. Schiavuta and R. Cini, Mar. Chem., 1999, 63, 235–253 CrossRef CAS.
- S. R. Piotrowicz, R. A. Duce, J. L. Fasching and C. P. Weisel, Mar. Chem., 1979, 7, 307–324 CrossRef CAS.
- C. P. Weisel, R. A. Duce, J. L. Fasching and R. W. Heaton, J. Geophys. Res.: Atmos., 1984, 89, 1607–1618 CrossRef.
- V. H. Grassian, Atmos. Environ., 2009, 43, 4666–4667 CrossRef CAS PubMed.
- C. Luo, N. Mahowald, T. Bond, P. Y. Chuang, P. Artaxo, R. Siefert, Y. Chen and J. Schauer, Global Biogeochem. Cycles, 2008, 22, GB1012, DOI:10.1029/2007gb002964.
- S. Scheinhardt, K. Muller, G. Spindler and H. Herrmann, Atmos. Environ., 2013, 74, 102–109 CrossRef CAS PubMed.
- S. Nickovic, A. Vukovic, M. Vujadinovic, V. Djurdjevic and G. Pejanovic, Atmos. Chem. Phys., 2012, 12, 845–855 CAS.
- C. R. Usher, A. E. Michel and V. H. Grassian, Chem. Rev., 2003, 103, 4883–4940 CrossRef CAS PubMed.
- S. Nickovic, A. Vukovic and M. Vujadinovic, Atmos. Chem. Phys., 2013, 13, 9169–9181 CrossRef.
- A. Ito and L. Xu, Atmos. Chem. Phys., 2014, 14, 3441–3459 CAS.
- E. R. Sholkovitz, P. N. Sedwick, T. M. Church, A. R. Baker and C. F. Powell, Geochim. Cosmochim. Acta, 2012, 89, 173–189 CrossRef CAS PubMed.
- M. S. Johnson, N. Meskhidze, F. Solmon, S. Gasso, P. Y. Chuang, D. Gaiero, R. M. Yantosca, S. Wu, Y. Wang and C. Carouge, J. Geophys. Res.: Atmos., 2010, 115, D15202, DOI:10.1029/2009jd013311.
- A. Ito, Global Biogeochem. Cycles, 2013, 27, 1–10 CrossRef CAS.
- J. Guo, A. Tilgner, C. Yeung, Z. Wang, P. K. K. Louie, C. W. Y. Luk, Z. Xu, C. Yuan, Y.-L. Gao, S. Poon, H. Herrmann, S. Lee, K. S. Lam and T. Wang, Environ. Sci. Technol., 2014, 48, 1443–1450 CrossRef CAS PubMed.
- R. A. Zaveri, C. M. Berkowitz, F. J. Brechtel, M. K. Gilles, J. M. Hubbe, J. T. Jayne, L. I. Kleinman, A. Laskin, S. Madronich and T. B. Onasch, et al., J. Geophys. Res.: Atmos., 2010, 115, D12304 CrossRef.
- A. Stefansson, Environ. Sci. Technol., 2007, 41, 6117–6123 CrossRef CAS.
- M. Lim, K. Chiang and R. Amal, J. Photochem. Photobiol., A, 2006, 183, 126–132 CrossRef CAS PubMed.
- W. Feng and D. Nansheng, Chemosphere, 2000, 41, 1137–1147 CrossRef CAS.
- J. Wittmer, S. Bleicher and C. Zetzsch, J. Phys. Chem. A, 2015, 119, 4373–4385 CrossRef CAS PubMed.
- Venny, S. Y. Gan and N. K. Ng, Chem. Eng. J., 2012, 213, 295–317 CrossRef CAS PubMed.
- C. L. Yap, S. Y. Gan and N. K. Ng, Chemosphere, 2011, 83, 1414–1430 CrossRef CAS PubMed.
- J. A. Zazo, J. A. Casas, A. F. Mohedano, M. A. Gilarranz and J. J. Rodriguez, Environ. Sci. Technol., 2005, 39, 9295–9302 CrossRef CAS.
- V. K. Gupta, I. Ali, T. A. Saleh, A. Nayak and S. Agarwal, RSC Adv., 2012, 2, 6380–6388 RSC.
- K. E. Daumit, A. J. Carrasquillo, J. F. Hunter and J. H. Kroll, Atmos. Chem. Phys., 2014, 14, 10773–10784 CAS.
- C. K. Duesterberg and T. D. Waite, Environ. Sci. Technol., 2007, 41, 4103–4110 CrossRef CAS.
- M. Shiraiwa, K. Selzle and U. Poschl, Free Radical Res., 2012, 46, 927–939 CrossRef CAS PubMed.
- S. A. Gurgueria, J. Lawrence, B. Coull, G. G. K. Murthy and B. Gonzalez-Flecha, Environ. Health Perspect., 2002, 110, 749–755 CrossRef.
- T. A. Cahill, D. E. Barnes, N. J. Spada, J. A. Lawton and T. M. Cahill, Aerosol Sci. Technol., 2011, 45, 1123–1134 CrossRef CAS PubMed.
- C. Faiola, A. M. Johansen, S. Rybka, A. Nieber, C. Thomas, S. Bryner, J. Johnston, M. Engelhard, P. Nachimuthu and K. S. Owen, Aerosol Sci. Technol., 2011, 45, 1109–1122 CrossRef CAS PubMed.
- H. Shen and C. Anastasio, Atmos. Chem. Phys., 2011, 11, 9671–9682 CAS.
- H. Shen, A. I. Barakat and C. Anastasio, Atmos. Chem. Phys., 2011, 11, 753–765 CAS.
- H. Shen and C. Anastasio, Atmos. Environ., 2012, 46, 665–668 CrossRef CAS PubMed.
- A. Leifer, The Kinetics of Environmental Aquatic Photochemistry, American Chemical Society, Washington, DC, 1988 Search PubMed.
- M. Moonshine, Y. Rudich, S. Katsman and E. R. Graber, Geophys. Res. Lett., 2008, 35, L20807 CrossRef.
- A. M'hemdi, B. Dbira, R. Abdelhedi, E. Brillas and S. Ammar, Clean: Soil, Air, Water, 2012, 40, 878–885 CrossRef PubMed.
- J. Arana, E. T. Rendon, J. M. Rodriguez, J. A. Melian, O. G. Diaz and J. P. Pena, Chemosphere, 2001, 44, 1017–1023 CrossRef CAS.
- S. G. Huber, G. Kilian and H. F. Scholer, Environ. Sci. Technol., 2007, 41, 7802–7806 CrossRef CAS.
- S. G. Huber, K. Kotte, H. F. Scholer and J. William, Environ. Sci. Technol., 2009, 43, 4934–4939 CrossRef CAS.
- S. Studenroth, S. G. Huber, K. Kotte and H. F. Scholer, Environ. Sci. Technol., 2013, 47, 1323–1329 CAS.
- H. Kipton, J. Powell and M. C. Taylor, Aust. J. Chem., 1982, 35, 739–756 CrossRef CAS.
- R. C. Hider, Z. D. Liu and H. H. Khodr, Metal chelation of polyphenol, in Methods in enzymology, ed. L. Packer, Academic Press, 2001, vol. 335, pp. 190–203 Search PubMed.
- M. J. Hynes and M. O. Coinceanainn, J. Inorg. Biochem., 2001, 85, 131–142 CrossRef CAS.
- J. M. Santana-Casiano, M. Gonzalez-Davila, A. G. Gonzalez and F. J. Millero, Aquat. Geochem., 2010, 16, 467–482 CrossRef CAS.
- D. E. Wheeler, J. H. Rodriguez and J. K. McCusker, J. Phys. Chem. A, 1999, 103, 4101–4112 CrossRef CAS.
- M. Elhabiri, C. Carrer, F. Marmolle and H. Traboulsi, Inorg. Chim. Acta, 2007, 360, 353–359 CrossRef CAS PubMed.
- C. G. Pierpont and R. M. Buchanan, Coord. Chem. Rev., 1981, 38, 45–87 CrossRef CAS.
- K. N. Raymond, G. Muller and B. F. Matzanke, Top. Curr. Chem., 1984, 123, 49–102 CAS.
- S. Kobayashi and A. Makino, Chem. Rev., 2009, 109, 5288–5353 CrossRef CAS PubMed.
- C. Xu, K. Xu, H. Gu, R. Zheng, H. Liu, X. Zhang, Z. Guo and B. Xu, J. Am. Chem. Soc., 2004, 126, 9938–9939 CrossRef CAS PubMed.
- S. Zurcher, D. Wackerlin, Y. Bethuel, B. Malisova, M. Textor, S. Tosatti and K. Gademann, J. Am. Chem. Soc., 2009, 128, 1064–1065 CrossRef PubMed.
- A. R. Statz, R. J. Meagher, A. E. Barron and P. B. Messersmith, J. Am. Chem. Soc., 2005, 127, 7972–7973 CrossRef CAS PubMed.
- R. C. Hider, A. R. Mohd-Nor, J. Silver, I. E. G. Morrison and L. V. C. Rees, J. Chem. Soc., Dalton Trans., 1981, 609–622 RSC.
- A. R. S. Ross, M. G. Ikonomou and K. J. Orians, Anal. Chim. Acta, 2000, 411, 91–102 CrossRef CAS.
- M.-A. Lutui, F. Gilard and M. Sablier, J. Mass Spectrom., 2008, 43, 1123–1131 CrossRef CAS PubMed.
- B. P. Heyden, E. J. Hauser, B. Mishra, G. A. Martinez, A. R. Bowie, T. Tyliszczak, T. N. Mtshali, A. N. Roychoudhury and S. C. B. Myneni, Environ. Sci. Technol. Lett., 2014, 1, 387–392 CrossRef.
- K. J. Schmalzl, C. M. Forsyth and P. D. Evans, Wood Sci. Technol., 1995, 29, 307–319 CAS.
- S. Hwang, C.-H. Lee and I.-S. Ahn, J. Ind. Eng. Chem., 2008, 14, 487–492 CrossRef CAS PubMed.
- D. R. Doerge, R. L. Divi and M. I. Churchwell, Anal. Biochem., 1997, 250, 10–17 CrossRef CAS PubMed.
- B. A. Poulin, J. N. Ryan and G. R. Aiken, Environ. Sci. Technol., 2014, 48, 10098–10106 CrossRef CAS PubMed.
- S. Baken, F. Degryse, L. Verheyen, R. Merckx and E. Smolders, Environ. Sci. Technol., 2011, 45, 2584–2590 CrossRef CAS PubMed.
- H. Fairbrother, F. M. Geiger, V. H. Grassian and J. C. Hemminger, Physical Chemistry of Environmental Interfaces, J. Phys. Chem. C, 2009, 113(6), 2035–2646 CAS.
- R. Signorell and A. K. Bertram, Physical Chemistry of Aerosols, Phys. Chem. Chem. Phys., 2009, 11, 7741–8104 RSC.
- V. H. Grassian, Surf. Sci., 2008, 602, 2955–2962 CrossRef CAS PubMed.
- S. G. Moussa, T. M. McIntire, M. Szori, M. Roeselová, D. J. Tobias, R. L. Grimm, J. C. Hemminger and B. J. Finlayson-Pitts, J. Phys. Chem. A, 2009, 113, 2060–2069 CrossRef CAS PubMed.
- G. Rubasinghege and V. H. Grassian, Chem. Commun., 2013, 49, 3071–3094 RSC.
- H. A. Al-Abadleh, H. A. Al-Hosney and V. H. Grassian, J. Mol. Catal. A: Chem., 2005, 228, 47–54 CrossRef CAS PubMed.
- D. B. Asay, A. L. Barnette and S. H. Kim, J. Phys. Chem. C, 2009, 113, 2128–2133 CAS.
- P. A. Thiel and T. E. Madey, Surf. Sci. Rep., 1987, 7, 211–385 CrossRef CAS.
- G. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed.
- B. R. Nichols, C. Rapa, V. Costa and R. Z. Hinrichs, J. Phys. Chem. C, 2009, 113, 2111–2119 CAS.
- F. G. Moore and G. L. Richmond, Acc. Chem. Res., 2008, 41, 739–748 CrossRef CAS PubMed.
- L. F. Scatena, M. G. Brown and G. L. Richmond, Science, 2001, 292, 908–912 CrossRef CAS PubMed.
- M. D. Cohen, R. C. Flagan and J. H. Seinfeld, J. Phys. Chem., 1987, 91, 4563–4574 CrossRef CAS.
- A. L. Goodman, E. T. Bernard and V. H. Grassian, J. Phys. Chem. A, 2001, 105, 6443–6457 CrossRef CAS.
- J. Baltrusaitis and V. H. Grassian, J. Phys. Chem. B, 2005, 109, 12227–12230 CrossRef CAS PubMed.
- L. A. Wijenayaka, G. Rubasinghege, J. Baltrusaitis and V. H. Grassian, J. Phys. Chem. C, 2012, 116, 12566–12577 CAS.
- X. Song and J.-F. Boily, Environ. Sci. Technol., 2013, 47, 7171–7177 CAS.
- X. Song and J.-F. Boily, Chem. Phys. Lett., 2013, 560, 1–9 CrossRef CAS PubMed.
- M. A. Donaldson, D. L. Bish and J. D. Raff, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18472–18477 CrossRef CAS PubMed.
- E. Kalman, T. Radnai, G. Palinkas, F. Hajdu and A. Vertes, Electrochim. Acta, 1988, 33, 1223–1228 CrossRef CAS.
- H. Ohtaki, Chem. Rev., 1993, 93, 1157–1204 CrossRef CAS.
- M. C. Pereira, L. C. A. Oliveira and E. Murad, Clay Miner., 2012, 47, 285–302 CrossRef CAS PubMed.
- J. F. Banfield and H. Zhang, Nanoparticles in the Environment, in Reviews in Mineralogy and Geochemistry, ed. J. F. Banfield and A. Navrotsky, Mineralogical Society of America, Washington, DC, 2001, vol. 44, pp. 1–58 Search PubMed.
- R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurrences, and Uses, Wiley-VCH, Weinheim, 2nd edn, 2003 Search PubMed.
- H. Gulley-Stahl, P. A. Hogan II, W. L. Schmidt, S. J. Wall, A. Buhrlage and H. A. Bullen, Environ. Sci. Technol., 2010, 44, 4116–4121 CrossRef CAS PubMed.
- Y. Yang, W. Yan and C. Jing, Langmuir, 2012, 28, 14588–14597 CrossRef CAS PubMed.
- L. A. Warren and E. A. Haack, Earth-Sci. Rev., 2001, 54, 261–320 CrossRef CAS.
- Z. B. Shi, M. D. Krom, T. D. Jickells, S. Bonneville, K. S. Carslaw, N. Mihalopoulos, A. R. Baker and L. G. Benning, Aeolian Res., 2012, 5, 21–42 CrossRef PubMed.
- G. Rubasinghege, R. W. Lentz, M. M. Scherer and V. H. Grassian, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6628–6633 CrossRef CAS PubMed.
- A. Philippe and G. E. Schaummann, Environ. Sci. Technol., 2014, 48, 8946–8962 CrossRef CAS PubMed.
- H. Liu, T. Chen and R. L. Frost, Chemosphere, 2014, 103, 1–11 CrossRef CAS PubMed.
- C. A. Lanzl, J. Baltrusaitis and D. M. Cwiertny, Langmuir, 2012, 28, 15797–15808 CrossRef CAS PubMed.
- Y. Yang, J. Duan and C. Jing, J. Phys. Chem. C, 2013, 117, 10597–10606 CAS.
- A. J. Anschutz and R. L. Penn, Geochem. Trans., 2005, 6, 60–66 CrossRef CAS PubMed.
- S. Orsetti, C. Laskov and S. B. Haderlein, Environ. Sci. Technol., 2013, 47, 14161–14168 CrossRef CAS PubMed.
- Z. Shi, J. M. Zachara, Z. Wang, L. Shi and J. K. Fredrickson, Geochim. Cosmochim. Acta, 2013, 121, 139–154 CrossRef CAS PubMed.
- Z. Shi, M. D. Krom, S. Bonneville and L. G. Benning, Environ. Sci. Technol., 2015, 49, 1472–1477 CrossRef CAS PubMed.
- P. J. DeMott, A. J. Prenni, X. Liu, S. M. Kreidenweis, M. D. Petters, C. H. Twohy, M. S. Richardson, T. Eidhammer and D. C. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11217–11222 CrossRef CAS PubMed.
- H. Chen and V. H. Grassian, Environ. Sci. Technol., 2013, 47, 10312–10321 CAS.
- M. Mochida, N. Umemoto, K. Kawamura, H.-J. Lim and B. J. Turpin, J. Geophys. Res.: Atmos., 2007, 112, D15209 CrossRef.
- A. H. Falkovich, G. Schkolnik, E. Ganor and Y. Rudich, J. Geophys. Res.: Atmos., 2004, 109, D02208 CrossRef.
- D. Jeong, K. Kim and W. Choi, Atmos. Chem. Phys., 2012, 12, 11125–11133 CrossRef CAS.
- K. Kim, W. Choi, M. R. Hoffmann, H. I. Yoon and B. K. Park, Environ. Sci. Technol., 2010, 44, 4142–4148 CrossRef CAS PubMed.
- T. Bartels-Rausch, H. W. Jacobi, T. F. Kahan, J. L. Thomas, E. S. Thomson, J. P. D. Abbatt, M. Ammann, J. R. Blackford, H. Bluhm, C. Boxe, F. Domine, M. M. Frey, I. Gladich, M. I. Guzman, D. Heger, T. Huthwelker, P. Klan, W. F. Kuhs, M. H. Kuo, S. Maus, S. G. Moussa, V. F. McNeill, J. T. Newberg, J. B. C. Pettersson, M. Roeselová and J. R. Sodeau, Atmos. Chem. Phys., 2014, 14, 1587–1633 CrossRef.
- V. F. McNeill, A. M. Grannas, J. P. D. Abbatt, M. Ammann, P. Ariya, T. Bartels-Rausch, F. Domine, D. J. Donaldson, M. I. Guzman, D. Heger, T. F. Kahan, P. Klan, S. Masclin, C. Toubin and D. Voisin, Atmos. Chem. Phys., 2012, 12, 9653–9678 CrossRef CAS.
- G. Rubasinghege, R. W. Lentz, H. Park, M. M. Scherer and V. H. Grassian, Langmuir, 2010, 26, 1524–1527 CrossRef CAS PubMed.
- D. M. Cwiertny, G. J. Hunter, J. M. Pettibone, M. M. Scherer and V. H. Grassian, J. Phys. Chem. C, 2009, 113, 2175–2186 CAS.
- C. J. Weschler, M. L. Mandich and T. E. Graedel, J. Geophys. Res., 1986, 91, 5189–5204 CrossRef CAS.
- R. Frank and W. Klöpffer, Chemosphere, 1988, 17, 985–994 CrossRef.
- P. Nissenson, D. Dabdub, R. Das, V. Maurino, C. Minero and D. Vione, Atmos. Environ., 2010, 44, 4859–4866 CrossRef CAS PubMed.
- A. E. Fazary, M. Taha and Y. H. Ju, J. Chem. Eng. Data, 2009, 54, 35–42 CrossRef CAS.
- T. D. Waite, Role of Iron in Light-Induced Environmetal Processes, in Environmental Photochemistry Part II, ed. P. Boule, D. W. Bahnemann and P. Robertson, Springer, Berlin, 2005, vol. 2M, pp. 225–298 Search PubMed.
- C. George, B. D'Anna, H. Herrmann, C. Weller, V. Vaida, D. J. Donaldson, T. Bartels-Rausch and M. Ammann, Emerging Areas in Atmospheric Photochemistry, in Atmospheric and Aerosol Chemistry, ed. V. F. McNeill and P. A. Ariya, Springer, Heidelberg, 2012, vol. 339, pp. 1–54 Search PubMed.
- B. C. Faust and R. G. Zepp, Environ. Sci. Technol., 1993, 27, 2517–2522 CrossRef CAS.
- H. B. Abrahamson, A. B. Rezvani and J. G. Brushmiller, Inorg. Chim. Acta, 1994, 226, 117–127 CrossRef CAS.
- E. M. Glebov, I. P. Pozdnyakov, V. P. Grivin, V. F. Plyusnin, X. Zhang, F. Wu and N. Deng, Photochem. Photobiol. Sci., 2011, 10, 425–430 CAS.
- C. Weller, S. Horn and H. Herrmann, J. Photochem. Photobiol., A, 2013, 268, 24–36 CrossRef CAS PubMed.
- C. Weller, S. Horn and H. Herrmann, J. Photochem. Photobiol., A, 2013, 255, 41–49 CrossRef CAS PubMed.
- S. V. Jovanovic, M. G. Simic, S. Steenken and Y. Hara, J. Chem. Soc., Perkin Trans. 2, 1998, 2365–2369 RSC.
- N. Quici and M. I. Litter, Photochem. Photobiol. Sci., 2009, 8, 975–984 CAS.
- N. Quici, M. I. Litter, A. M. Braun and E. Oliveros, J. Photochem. Photobiol., A, 2008, 197, 306–312 CrossRef CAS PubMed.
- F. J. Benitez, F. J. Real, J. L. Acero, A. I. Leal and C. Garcia, J. Hazard. Mater. B, 2005, 126, 31–39 CrossRef CAS PubMed.
- G. Lofrano, L. Rizzo, M. Grassi and V. Belgiorno, Desalination, 2009, 249, 878–883 CrossRef CAS PubMed.
- Y. Samet, I. Wali and R. Abdelhedi, Environ. Sci. Pollut. Res., 2011, 18, 1497–1507 CrossRef CAS PubMed.
- A. Machulek, C. Vautier-Giongo, J. E. F. Moraes, C. A. O. Nascimento and F. H. Quina, Photochem. Photobiol. Sci., 2006, 82, 208–212 CAS.
- J. Kiwi, A. Lopez and V. Nadtochenko, Environ. Sci. Technol., 2000, 34, 2162–2168 CrossRef CAS.
- H. Hasegawa and P. Neta, J. Phys. Chem., 1978, 82, 854–857 CrossRef.
- D. Vione, V. Maurino, C. Minero, P. Calza and E. Pelizzetti, Environ. Sci. Technol., 2005, 39, 5066–5075 CrossRef CAS.
- H. Herrmann, Chem. Rev., 2003, 103, 4691–4716 CrossRef CAS PubMed.
- G. V. Buxton, M. Bydder, G. A. Salmon and J. E. William, Phys. Chem. Chem. Phys., 2000, 2, 237–245 RSC.
- M. L. Alegre, M. Gerones, J. A. Rosso, S. G. Bertolotti, A. M. Braun, D. O. Martire and M. C. Gonzalez, J. Phys. Chem. A, 2000, 104, 3117–3125 CrossRef CAS.
- F. Laturnus, I. Fahimi, M. Gryndler, A. Hartmann, M. R. Heal, M. Matucha, H. F. Schöler, R. Schroll and T. Svensson, Environ. Sci. Pollut. Res., 2005, 12, 233–244 CrossRef CAS.
- F. Keppler, R. Borchers, J. T. G. Hamilton, G. Kilian, J. Pracht and H. F. Scholer, Environ. Sci. Technol., 2006, 40, 130–134 CrossRef CAS.
- K. W. Oum, M. J. Lakin, D. O. DeHaan, T. Brauers and B. J. Finlayson-Pitts, Science, 1998, 279, 74–77 CrossRef CAS.
- B. D. Finley and E. S. Saltzman, J. Geophys. Res.: Atmos., 2008, 113, D21301 CrossRef.
- B. D. Finley and E. S. Saltzman, Geophys. Res. Lett., 2006, 33, L11809 CrossRef.
- D. I. Resser, C. George and D. J. Donaldson, J. Phys. Chem. A, 2009, 113, 8591–8595 CrossRef PubMed.
- A. Jammoul, S. Dumas, B. D'Anna and C. George, Atmos. Chem. Phys., 2009, 9, 4229–4237 CrossRef CAS.
- J. Ofner, N. Blazer, J. Buxmann, H. Grothe, P. Schmitt-Kopplin, U. Platt and C. Zetzsch, Atmos. Chem. Phys., 2012, 12, 5787–5806 CrossRef CAS.
- L. Deguillaume, M. Leriche, K. Desboeufs, G. Mailhot, C. George and N. Chaumerliac, Chem. Rev., 2005, 105, 3388–3431 CrossRef CAS PubMed.
- P. Nissenson, C. J. H. Knox, B. J. Finlayson-Pitts, L. F. Phillips and D. Donald, Phys. Chem. Chem. Phys., 2006, 8, 4700–4710 RSC.
- F. Karagulian, C. W. Dilbeck and B. J. Finlayson-Pitts, J. Phys. Chem. A, 2009, 113, 7205–7212 CrossRef CAS PubMed.
- M. Ndour, B. D'Anna, C. George, O. Ka, Y. Balkanski, J. Kleffmann, K. Stemmler and M. Ammann, Geophys. Res. Lett., 2008, 35, L05812 CrossRef.
- M. Brigante, D. Cazoir, B. D'Anna, C. George and D. J. Donaldson, J. Phys. Chem. A, 2008, 112, 9503–9508 CrossRef CAS PubMed.
- D. I. Resser, A. Jammoul, D. Clifford, M. Brigante, B. D'Anna, C. George and D. J. Donaldson, J. Phys. Chem. A, 2009, 113, 2071–2077 CrossRef PubMed.
- M. E. Monge, T. Rosenorn, O. Favez, M. Muller, G. Adler, A. A. Riziq, Y. Rudich, H. Herrmann, C. George and B. D'Anna, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6840–6844 CrossRef CAS PubMed.
- A. Fujishima, M. Saeki, N. Kimizuka, M. Taniguchi and S. Suga, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 7318–7333 CrossRef.
- H. Zhang, M. Bayne, S. Fernando, B. Legg, M. Zhu, R. L. Penn and J. F. Banfield, J. Phys. Chem. C, 2011, 115, 17704–17710 CAS.
- A. I. Martinez, M. A. Garcia-Lobato and D. L. Perry, Study of the Properties of Iron Oxide Nanostructures, in Research in Nanotechnology Developments, ed. A. Barranon, Nova Science Publishers, Inc., New York, 2009, pp. 183–194 Search PubMed.
- G. Rubasinghege, S. Elzey, J. Baltrusaitis, P. M. Jayaweera and V. H. Grassian, J. Phys. Chem. Lett., 2010, 1, 1729–1737 CrossRef CAS.
- C. Zwiener and F. H. Frimmel, Anal. Bioanal. Chem., 2004, 378, 862–874 CrossRef CAS PubMed (review).
- A. R. Baker and P. L. Croot, Mar. Chem., 2010, 120, 4–13 CrossRef CAS PubMed.
- H. Chen, C. E. Nanayakkara and V. H. Grassian, Chem. Rev., 2012, 112, 5919–5948 CrossRef CAS PubMed.
- H. Chen, J. G. Navea, M. A. Young and V. H. Grassian, J. Phys. Chem. A, 2011, 115, 490–499 CrossRef CAS PubMed.
- B. Xu, T. Zhu, X. Tang and J. Shang, Sci. China: Chem., 2010, 53, 2644–2651 CrossRef CAS.
- M. Sassine, L. Burel, B. D'Anna and C. George, Atmos. Environ., 2010, 44, 5468–5475 CrossRef CAS PubMed.
- S. A. Styler and D. J. Donaldson, Environ. Sci. Technol., 2011, 45, 10004–10012 CrossRef CAS PubMed.
- F. Rifkha Kameel, F. Riboni, M. R. Hoffmann, S. Enami and A. J. Colussi, J. Phys. Chem. C, 2014, 118, 29151–29158 Search PubMed.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, Taylor & Francis, Boca Raton, 2012-2013, vol. 93 Search PubMed.
- E. Mentasti and E. Pelizzetti, J. Chem. Soc., Dalton Trans., 1973, 2605–2608 RSC.
- R. Oder and D. A. Goring, Spectrochim. Acta, 1971, 27A, 2285–2288 CrossRef.
- S. I. Ikawa and S. Maeda, Spectrochim. Acta, 1968, 24A, 655–665 CrossRef.
- G. C. Pimentel and A. L. McClellan, The Hydrogen Bond, W.H. Freeman and Co., San Fransisco, 1960 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |