
Magnetic nano-Fe3O4@TiO2/Cu2O core–shell composite: an efficient novel catalyst for the regioselective synthesis of 1,2,3-triazoles using a click reaction
Abstract
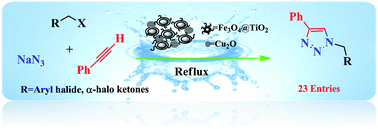
A magnetic nano-Fe3O4@TiO2/Cu2O core–shell composite, optimized for the loading amount of Cu2O, was successfully prepared and fully characterized by analyzing FT-IR, XRD, XPS, FEG-SEM, EDS, and VSM data. The catalytic activity of the nano-Fe3O4@TiO2/Cu2O core–shell composite was examined in a classical one-pot and three-component reaction of terminal alkynes, benzyl halides, and sodium azide in water that was observed to proceed smoothly and completed in good yields with high regioselectivity. The catalyst was conventionally recovered using an external magnet and was reused in at least five successive runs without an appreciable loss of activity.
 Please wait while we load your content...
Please wait while we load your content...

