DOI:
10.1039/C5RA02972D
(Paper)
RSC Adv., 2015,
5, 25642-25649
Ultrasound-assisted regio- and stereoselective synthesis of bis-[1′,4′-diaryl-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] derivatives via 1,3-dipolar cycloaddition†
Received
16th February 2015
, Accepted 4th March 2015
First published on 4th March 2015
Abstract
The 1,3-dipolar cycloaddition reaction of the bis-hydrazonoyl chlorides with 2-arylidene-1-benzosuberone derivatives afforded the corresponding bis-[1′,4′-diaryl-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] derivatives. The reaction is carried out under ultrasonic irradiation as well as under conventional heating. The factors affecting the optimization of the cycloaddition reaction are examined in detail. X-ray crystallographic analysis was used in the establishment of the regio- and stereochemistry of the cycloaddition products.
Introduction
Ultrasound, as an environmentally sustainable technique, has found several applications in synthetic organic chemistry, in materials science, in medicinal chemistry and in life sciences.1–5 Compared with conventional heating, the key advantages of ultrasound irradiation are: (1) increasing the yields, (2) decreasing the reaction time and (3) increasing the products' purity and selectivity.6,7 1,3-Dipolar cycloaddition is one of the most valuable synthetic routes for the synthesis of five-membered heterocycles.8,9 Spiropyrazoline derivatives are involved in several pharmaceutical agents, such as antiviral and antibacterial activities,10,11 anti-cancer agents,12 as well as acetyl-CoA carboxylase inhibiting activities.13 Benzosuberone moiety (6,7,8,9-tetrahydrobenzo-cyclohepten-5-one) was found in variety of biologically active compounds that proved to have anti-cancer,14–16 anti-proliferative17 and antiangiogenic agents18,19 as well as human aminopeptidase20,21 and acetylcholinesterase inhibiting activities.22 In continuation of our research work on the [3 + 2] and [4 + 1] cycloadditions towards pyrazole synthesis and on the chemistry of bis-hydrazonoyl chlorides 1a–c,23–31 we report here their first example of ultrasound-assisted 1,3-dipolar cycloaddition with 2-arylidene-1-benzosuberones 3a–g for the construction of the bis-[1-oxo-spiro-benzosuberane-2,5′-pyrazoline] derivatives 4a–m. The regio- and stereoselectivity of the cycloaddition mode will be examined by measuring the X-ray crystallographic analysis of the products.
Results and discussion
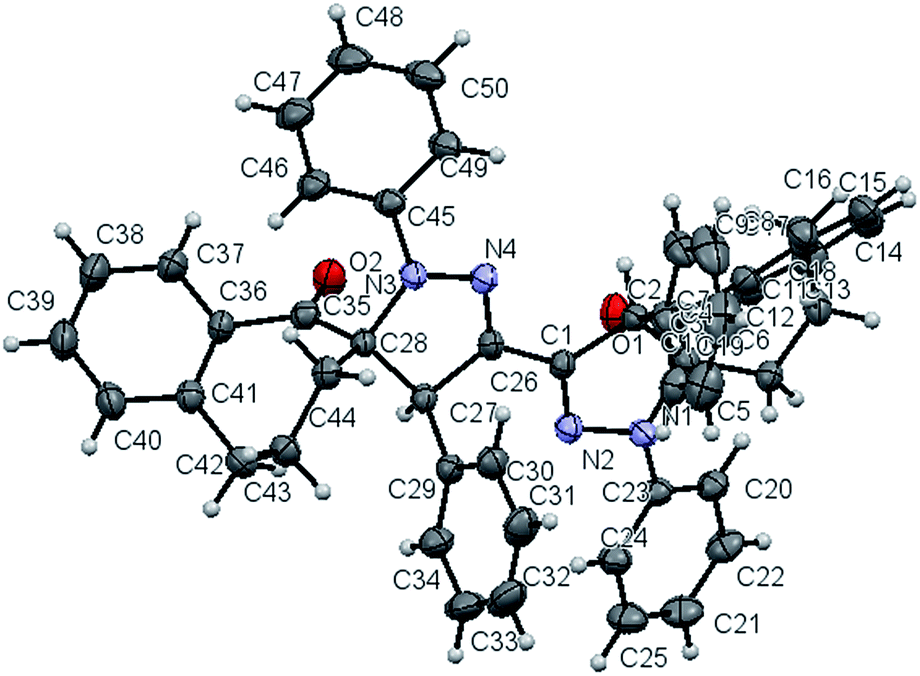
Effect of parameters like choice of solvent, type of the used base, mode of activation (ultrasonic irradiation and conventional heating) will be extensively examined. Firstly, using triethylamine (TEA) as a base, the effect of solvents (ethanol, benzene, toluene and chloroform) on the 1,3-dipolar cycloaddition reactions of the bis-nitrilimine 2a, [generated in situ from the bis-hydrazonoyl chloride 1a with base], with 2-benzylidene-1-benzosuberone (3a), as a model reaction, was evaluated under ultrasound irradiation and conventional heating and the results are listed in Table 1. Thus, when the 1,3-dipolar cycloaddition of 2a with 3a using EtOH–Et3N under ultrasonic irradiation at 70 °C (110 Watt), the starting substrates were completely consumed after 3 hours to afford only one isolable product. The HRMS of the obtained pure product showed a peak at m/z 730.3302, this value corresponds to the molecular formula C50H42N4O2. This result declared that the cycloaddition reaction proceeded between 2a and 3a in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio. Since the bis-nitrilimine 2a has two possible 1,3-dipole attacking sites, there are three expected cycloadducts for which structures 4a, 5a and 6a can be assigned (Scheme 1). The 1H NMR spectrum of the reaction product revealed sets of multiplets in the region δ 1.0–2.94 due to the aliphatic CH2's protons and a singlet signal at δ 4.97 in addition to aromatic protons signals in the region δ 6.72–7.51. The singlet at δ 4.97 is due to the pyrazoline-4H proton which is close to analogously reported pyrazoline-4H proton at δ ≈ 5 (ref. 23 and 32) and not consistent with the pyrazoline-5H proton which appears at δ value >5.6.33 These data support the bis-[1′,4′-diphenyl-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] structure (4a) and rules out the other regioisomeric structures 5a or 6a (Scheme 1). Further, the 13C NMR spectrum showed five sp3 carbon-signals in the region δ 21.8–79.4 among them a peak at 79.35 due to spiropyrazoline-5-C atom which is close to analogous reported simple structures.32 The regio- and stereoselective formation of structure 4a was unequivocally evidenced by measuring its X-ray single crystal analysis (Fig. 1).34
:2 molar ratio. Since the bis-nitrilimine 2a has two possible 1,3-dipole attacking sites, there are three expected cycloadducts for which structures 4a, 5a and 6a can be assigned (Scheme 1). The 1H NMR spectrum of the reaction product revealed sets of multiplets in the region δ 1.0–2.94 due to the aliphatic CH2's protons and a singlet signal at δ 4.97 in addition to aromatic protons signals in the region δ 6.72–7.51. The singlet at δ 4.97 is due to the pyrazoline-4H proton which is close to analogously reported pyrazoline-4H proton at δ ≈ 5 (ref. 23 and 32) and not consistent with the pyrazoline-5H proton which appears at δ value >5.6.33 These data support the bis-[1′,4′-diphenyl-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] structure (4a) and rules out the other regioisomeric structures 5a or 6a (Scheme 1). Further, the 13C NMR spectrum showed five sp3 carbon-signals in the region δ 21.8–79.4 among them a peak at 79.35 due to spiropyrazoline-5-C atom which is close to analogous reported simple structures.32 The regio- and stereoselective formation of structure 4a was unequivocally evidenced by measuring its X-ray single crystal analysis (Fig. 1).34
Table 1 Optimization of the reaction conditions for synthesis of 4a
| Entry |
Solvent |
Base |
Yield% of 4aa |
| )))d [3 h] |
Δe [36 h] |
| Reaction conditions: bis-hydrazonoyl chloride 1a (2 mmol), 2-benzylidene-1-benzosuberone 3a (4 mmol), base (4 mmol) and solvent (25 mL) under sonication (at 70 °C for 3 h) or conventional heating (at reflux for 36 h). Benzosuberone derivative 3a was detected. Benzosuberone derivative 3a was completely recovered. ))) = ultrasonic irradiation. Δ = conventional heating. |
| 1 |
Ethanol |
TEA |
87 |
35b |
| 2 |
Benzene |
TEA |
13b |
11b |
| 3 |
Toluene |
TEA |
19b |
15b |
| 4 |
Chloroform |
TEA |
Tracec |
Tracec |
| 5 |
Ethanol |
DABCO |
56 |
29b |
| 6 |
Ethanol |
DBU |
44b |
25b |
| 7 |
Ethanol |
CsF |
Tracec |
Tracec |
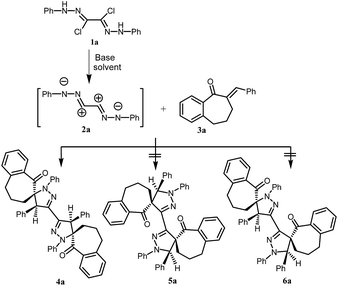 |
| | Scheme 1 Regio- and stereoselective synthesis of bis-[1′,4′-diphenyl-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] (4a). | |
 |
| | Fig. 1 ORTEP plot of the X-ray crystallographic data determined for 4a. | |
The yield of the product 4a under ultrasound irradiation was found to be 87% after 3 hours, however, when the same cycloaddition reaction was repeated under conventional heating at reflux temperature using EtOH–Et3N, the reaction completed after 36 hours but with sharp decrease in the isolated yield (35%) (Table 1, entry 1). When the same reaction between 2a and 3a was repeated using benzene instead of ethanol in the presence of Et3N, the yield of the product 4a was very poor either under conventional heating (11% after 36 h) or ultrasound irradiation (13% after 3 h) (Table 1, entry 2) and the substrate 3a was incompletely reacted. Further, toluene was also not appropriate solvent where the yield of 4a was 15% after 36 h of conventional heating and 19% after 3 h of ultrasound irradiation, with incomplete reaction of the substrate 3a. No reaction at all took place either under conventional heating or ultrasound irradiation when chloroform was used as a solvent, where the starting substrate 3a was completely recovered. Therefore, ethanol is the solvent of choice for conducting this 1,3-dipolar cycloaddition reaction under ultrasonic condition.
Next, using ethanol as reaction solvent, the effect of further organic and inorganic bases (DABCO, DBU and CsF) on the behaviour of this cycloaddition reaction was also evaluated. Thus, when EtOH–DABCO system was applied, compound 3a was isolated in 56% yield (after 3 h of ultrasound irradiation) and in 29% yield (after 36 h of conventional heating) (Table 1, entry 5). The use of EtOH–DBU resulted in the formation of compound 4a in 44% yield (after 3 h of ultrasound irradiation) and in 25% yield (after 36 h of conventional heating) (Table 1, entry 6). The inorganic base CsF was not appropriate the cycloaddition of 1a with 3a where only traces of 4a was detected by TLC both under conventional heating and ultrasound irradiation and the starting substrate 3a was almost completely recovered (Table 1, entry 7).
Under the optimized reaction conditions above, we investigated the substrate scope of this reaction as shown in Scheme 2. The methodology was found to be applicable to a range of the substrates; 2-arylidene-1-benzosuberone derivatives 3a–g and bis-hydrazonoyl chlorides 1a–c. Thus, the regioselective 1,3-dipolar cycloaddition reaction of the bis-nitrilimine 2a with the 2-arylidene-1-benzosuberone derivatives 3a–g was conducted using EtOH–Et3N under both ultrasonic irradiation and conventional heating and afforded the corresponding bis-[1′,4′-diaryl-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] derivatives 4a–g, Scheme 2. The yields of the cycloadducts 4a–g varied between 76 and 91% (after 3 h of sonication) and between 25 and 42% yields (after 36 h of conventional heating) as shown in Table 2, entries 1–7. Similar to compound 4a, the regio- and stereoselective formation of the bis-[1′,4′-diaryl-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] structures 4b–g were determined from their full spectral data (IR, HRMS, 1H and 13C NMR spectra) and by measuring the single crystal X-ray analyses of compounds 4b–d as depicted in Fig. 2–4.34
 |
| | Scheme 2 Regio- and stereoselective synthesis of the bis-spiropyrazoline derivatives 4a–m. | |
Table 2 1,3-Dipolar cycloaddition reactions of 1a–c with 3-arylidenebenzosuberones 3a–g
| Entry |
Reactants |
Products |
Ar1 |
Ar2 |
Yield% 4a–ma |
| )))b [3 h] |
Δc [36 h] |
| Reaction conditions: bis-hydrazonoyl chlorides 1a–c (2 mmol), 2-arylidene-1-benzosuberones 3a–g (4 mmol), Et3N (4 mmol) and EtOH (25 mL) under sonication (at 70 °C for 3 h) or conventional heating (at reflux for 36 h). Benzosuberone derivatives 3a–g were completely consumed. Benzosuberone derivatives 3a–g were detected by TLC. |
| 1 |
1a + 3a |
4a |
C6H5 |
C6H5 |
87 |
35 |
| 2 |
1a + 3b |
4b |
C6H5 |
4-ClC6H4 |
91 |
42 |
| 3 |
1a + 3c |
4c |
C6H5 |
2-ClC6H4 |
85 |
33 |
| 4 |
1a + 3d |
4d |
C6H5 |
4-NO2C6H4 |
86 |
32 |
| 5 |
1a + 3e |
4e |
C6H5 |
4-MeOC6H4 |
79 |
29 |
| 6 |
1a + 3f |
4f |
C6H5 |
2,4-(MeO)2C6H3 |
76 |
25 |
| 7 |
1a + 3g |
4g |
C6H5 |
4-MeC6H4 |
80 |
36 |
| 8 |
1b + 3a |
4h |
4-ClC6H4 |
C6H5 |
83 |
38 |
| 9 |
1b + 3b |
4i |
4-ClC6H4 |
4-ClC6H4 |
88 |
45 |
| 10 |
1b + 3f |
4j |
4-ClC6H4 |
2,4-(MeO)2C6H3 |
84 |
41 |
| 11 |
1c + 3a |
4k |
4-MeC6H4 |
C6H5 |
90 |
37 |
| 12 |
1c + 3b |
4l |
4-MeC6H4 |
4-ClC6H4 |
87 |
51 |
| 13 |
1c + 3f |
4m |
4-MeC6H4 |
2,4-(MeO)2C6H3 |
72 |
22 |
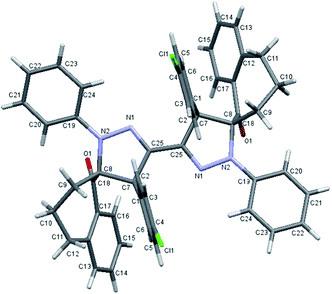 |
| | Fig. 2 The X-ray crystallographic data determined for 4b. | |
 |
| | Fig. 3 ORTEP plot of the X-ray crystallographic data determined for 4c. | |
 |
| | Fig. 4 ORTEP plot of the X-ray crystallographic data determined for 4d. | |
To further expand and to generalize the highly regio- and stereoselective behavior of the cycloaddition reaction as examined between the bis-nitrilimine 2a and dipolarophiles 3a–g, the 1,3-dipolar cycloaddition reaction of the bis-nitrilimine 2b with the dipolarophiles 3a,b,f was similarly conducted and the data are outlined in Table 2, entries 8–10. Thus, ultrasonic irradiation of the bis-nitrilimine 2b with the dipolarophiles 3a,b,f in ethanol in the presence of Et3N afforded the desired bis-[1-oxo-spiro-benzosuberane-2,5′-pyrazoline] derivatives 4h,i,j in 83, 88 and 84% isolated yields, respectively. The same compounds 4h,i,j were obtained from the reaction of 2b with 3a,b,f in 38, 45 and 41% yields, respectively, when refluxed under conventional heating for 36 h in ethanol in the presence of Et3N. Structures 4h,i,j were established from their spectral data (IR, HRMS, 1H and 13C NMR spectra) as shown in the experimental section.
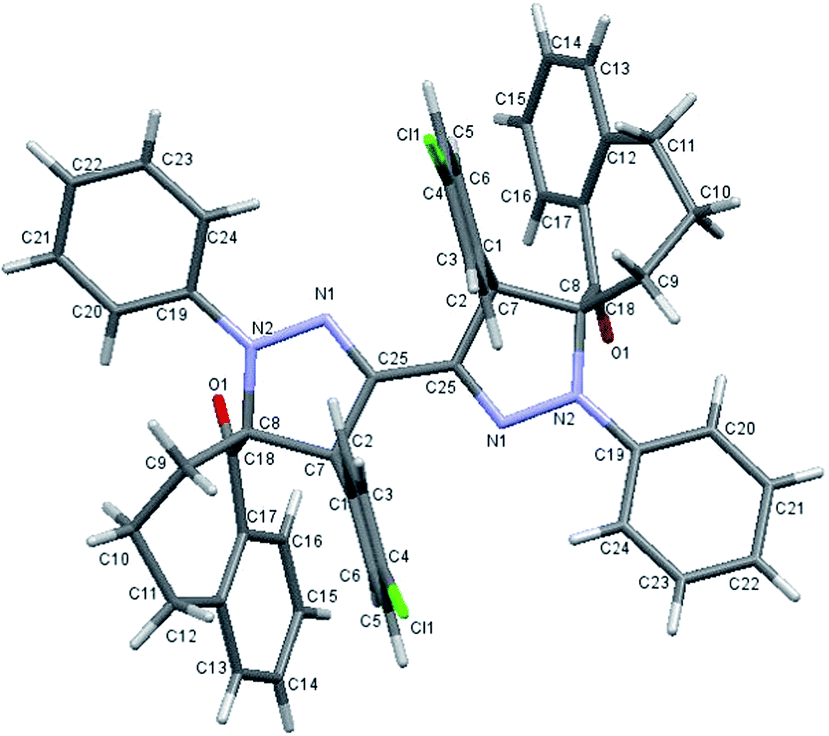
Finally, the regio- and stereoselective 1,3-dipolar cycloaddition reaction of the bis-nitrilimine 2c and dipolarophiles 3a,b,f, was similarly performed. Thus, carrying out the reaction of 2c with 3a,b,f in ethanol solvent and Et3N as base resulted in the formation of the bis-[1-oxo-spiro-benzosuberane-2,5′-pyrazoline] derivatives 4k,l,m in 90, 87 and 72% yields, respectively (under ultrasonic irradiation) and in 37, 51 and 22% yields, respectively (under conventional heating) as shown in Table 2, entries 11–13. The single crystal X-ray analysis of compound 4l showed in Fig. 5,34 provided a firm support the for the regio- and stereoselective manner of the cycloaddition process.
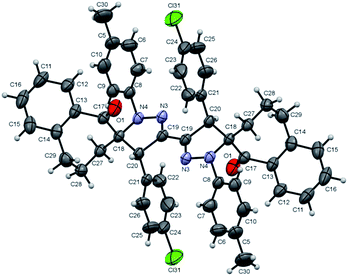 |
| | Fig. 5 ORTEP plot of the X-ray crystallographic data determined for 4l. | |
Experimental
General
Melting points were recorded on a Griffin melting point apparatus and are reported uncorrected. IR spectra were recorded using KBr disks using a Perkin-Elmer System 2000 FT-IR spectrophotometer. 1H-NMR (400 MHz) or (600 MHz) and 13C-NMR (100 MHz) or (150 MHz) spectra were recorded at 25 °C using CDCl3 or DMSO-d6 as solvent with TMS as internal standard on a Bruker DPX 400 or 600 super-conducting NMR spectrometer. Chemical shifts are reported in ppm. Low-resolution electron impact mass spectra [MS (EI)] and High-resolution electron impact mass spectra [HRMS (EI)] were performed on high resolution GC-MS (DFS) thermo spectrometers at 70.1 eV using magnetic sector mass analyzer. Follow up of the reactions and checking homogeneity of the prepared compounds was made by thin layer chromatography (TLC). The crystal structures were determined by a Rigaku R-AXIS RAPID diffractometer and Bruker X8 Prospector and the single crystal data collections were made by using Cu-Kα radiation. The data were collected at room temperature. The structure was solved by direct methods and was expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. The structure was solved and refined using the Bruker SHELXTL Software Package (structure solution program – SHELXS-97 and refinement program – SHELXL-97).35 Data were corrected for the absorption effects using the multi-scan method (SADABS). Sonication was performed in MKC6, Guyson ultrasonic bath (model-MKC6, operating frequency 38 kHz ± 10% and an output power of 110 Watts) with digital timer (6 s to 100 min) and heater allows solution heating to be set from 20 °C to 80 °C in 1 °C increments. The inside tank dimensions are 150 × 300 × 150 mm (length × width × depth) with a fluid capacity of 6 liters. 2-Arylidene-1-benzosuberone derivatives 3a–g was prepared according to the literature procedures.36
N,N′-Diphenylethane(bis-hydrazonoyl dichloride) (1a). Yield: (76%); recrystallized from acetic acid as yellowish white crystals; mp. 190–191 °C (Lit. mp.37 188–190 °C); 1H-NMR (DMSO-d6): δ = 6.86–6.91 (m, 2H, Ar-H), 7.25–7.33 (m, 8H, Ar-H), 10.36 (s, 2H, NH); 13C-NMR (DMSO-d6): δ = 113.63, 117.74, 120.96, 129.12, 143.65.
N,N′-Di-(4-chlorophenyl)ethane(bis-hydrazonoyl dichloride) (1b). Yield: (77%); recrystallized from acetic acid as luster buff crystals; mp. 224–225 °C (Lit. mp.38 223–225 °C); 1H-NMR (DMSO-d6): δ = 7.31–7.32 (m, 8H, Ar-H), 10.34 (s, 2H, NH); 13C-NMR (DMSO-d6): δ = 115.16, 118.39, 124.51, 128.98, 142.58.
N,N′-Di-(4-tolyl)ethane(bis-hydrazonoyl dichloride) (1c). Yield: (80%); recrystallized from acetic acid as luster pale yellow crystals, mp. 199–200 °C (Lit. mp.38 198–200 °C); 1H-NMR (DMSO-d6): δ = 2.24 (s, 6H, 2CH3), 7.10 (d, J = 7.8 Hz, 4H, Ar-H), 7.22 (d, J = 7.8 Hz, 4H, Ar-H), 10.05 (s, 2H, NH); 13C-NMR (DMSO-d6): δ = 20.30, 113.60, 117.22, 129.52, 129.61, 141.42.
Synthesis of bis-[1′,4′-diaryl-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] 4a–m.
General method A. To a mixture of the appropriate bis-hydrazonoyl chloride 1a–c (2 mmol) and the appropriate 2-arylidene-1-benzosuberone derivative 3a–g (4 mmol each) in the appropriate dry solvent (ethanol, benzene, toluene or chloroform) (25 mL), triethylamine (0.6 mL, 4 mmol) was added portion-wise. The mixture was heated at refluxing temperature and the reaction was followed up by TLC and continued for 36 h, then left to cool to room temperature. The solvent was removed, in each case, under reduced pressure and the residue was triturated with methanol to give yellow or pale-brown colored products. The solid products that formed were filtered off, washed with ethanol, dried and recrystallized from dimethylformamide (DMF)/ethanol (3:1) to afford the corresponding bis-[1′,4′-diaryl-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] derivatives 4a–m as pure products.
General method B. To a mixture of the appropriate bis-hydrazonoyl chloride 1a–c (2 mmol) and the appropriate 2-arylidene-1-benzosuberone derivative 3a–g (4 mmol each) in the appropriate dry solvent (ethanol, benzene, toluene or chloroform) (25 mL), triethylamine (0.6 mL, 4 mmol) was added portion-wise. The reaction mixture was sonicated in MKC6, Guyson ultrasonic bath (Model-MKC6, operating frequency 38 kHz ± 10% and an output power of 110 Watts) for 3 h at 70 °C. The reaction was controlled by TLC and continued until the starting substrates were completely consumed, then left to cool to room temperature. The solid products that formed were filtered off, washed with ethanol, dried and recrystallized from dimethylformamide (DMF)/ethanol (3:1) to afford the corresponding bis-[1′,4′-diaryl-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] 3a–m as pure products.
Bis-[1′,4′-diphenyl-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] (4a). As yellow crystals, mp. 270–271 °C; IR (KBr): ν/cm−1 1690 (CO); 1H-NMR (DMSO-d6): δ = 1.00–1.05 (m, 2H, H of CH2), 1.36–1.41 (m, 2H, H of CH2), 1.65–1.69 (m, 2H, H of CH2), 2.29–2.35 (m, 2H, H of CH2), 2.73–2.77 (m, 2H, H of CH2), 2.91–2.94 (m, 2H, H of CH2), 4.97 (s, 2H, pyrazole-CH), 6.72 (d, J = 7.8 Hz, 4H, Ar-H), 6.88 (t, J = 7.8 Hz, 4H, Ar-H), 7.14 (t, J = 7.2 Hz, 4H, Ar-H), 7.27 (t, J = 7.8 Hz, 4H, Ar-H), 7.31–7.38 (m, 10H, Ar-H) and 7.51 ppm (t, J = 7.8 Hz, 2H, Ar-H); 13C-NMR (DMSO-d6): δ = 21.84, 27.97, 32.07 (CH2), 58.91 (pyrazole-CH), 79.35 (spiro-C), 118.20, 121.52, 126.93, 127.76, 128.17, 128.48, 128.70, 129.22, 129.27, 132.46, 136.35, 137.68, 139.24, 142.46, 144.16 and 205.19 ppm (Ar-C and CO); MS (EI): m/z (%) 732 (M+ + 2, 13.45), 731 (M+ + 1, 46.83), 730 (M+, 77.05); HRMS (EI): m/z calcd for C50H42N4O2 (M+) 730.3302, found: 730.3302. Crystal data, C50H42N4O2, M = 730.92, monoclinic, a = 11.0471(4) Å, b = 18.9631(7) Å, c = 22.9273(8) Å, V = 4728.9(3) Å3, α = γ = 90°, β = 100.077(2)°, space group: P121/c1, Z = 4, Dcalc = 1.232 g cm−3, no. of reflection measured 53472, θmax = 66.71°, R1 = 0.0581.34
Bis-[1′-phenyl-4′-(4-chlorophenyl)-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] (4b). As yellow crystals, mp. 277–278 °C; IR (KBr): ν/cm−1 1686 (CO); 1H-NMR (DMSO-d6): δ = 0.93–1.01 (m, 2H, H of CH2), 1.38–1.46 (m, 2H, H of CH2), 1.69–1.74 (m, 2H, H of CH2), 2.31–2.37 (m, 2H, H of CH2), 2.72–2.76 (m, 2H, H of CH2), 2.94–3.00 (m, 2H, H of CH2), 5.09 (s, 2H, pyrazole-CH), 6.75 (d, J = 8.0 Hz, 4H, Ar-H), 6.89–6.95 (m, 4H, Ar-H), 7.14–7.22 (m, 8H, Ar-H), 7.27 (d, J = 7.6 Hz, 2H, Ar-H), 6.35 (t, J = 7.6 Hz, 2H, Ar-H), 7.42 (d, J = 8.0 Hz, 4H, Ar-H) and 7.52 ppm (t, J = 7.6 Hz, 2H, Ar-H); 13C-NMR (DMSO-d6): δ = 22.13, 27.86, 31.88 (CH2), 57.95 (pyrazole-CH), 79.21 (spiro-C), 118.39, 121.90, 126.97, 128.36, 128.58, 129.29, 130.81, 132.43, 132.68, 135.68, 137.75, 139.14, 142.46, 142.92, 144.06 and 205.10 ppm (Ar-C and CO); MS (EI): m/z (%) 800 (M+ + 2, 44.95), 799 (M+ + 1, 29.46), 798 (M+, 55.89); HRMS (EI): m/z calcd for C50H40Cl2N4O2 (M+) 798.2523, found 798.2523. Crystal data, C50H40Cl2N4O2, M = 799.76, orthorhombic, a = 22.2297(8) Å, b = 11.6884(5) Å, c = 15.5606(6) Å, V = 4043.1(3) Å3, α = β = γ = 90°, space group: Pcca, Z = 4, Dcalc = 1.314 g cm−3, no. of reflection measured 12902, θmax = 66.62°, R1 = 0.0498.34
Bis-[1′-phenyl-4′-(2-chlorophenyl)-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] (4c). As yellow crystals, mp. >300 °C; IR (KBr): ν/cm−1 1687 (CO); 1H-NMR (DMSO-d6): δ = 1.21–1.27 (m, 2H, H of CH2), 1.50–1.56 (m, 2H, H of CH2), 1.93–1.97 (m, 2H, H of CH2), 2.34–2.39 (m, 2H, H of CH2), 2.76–2.86 (m, 4H, 2H of CH2), 5.29 (s, 2H, pyrazole-CH), 6.52 (d, J = 7.6 Hz, 2H, Ar-H), 6.79 (d, J = 8.0 Hz, 4H, Ar-H), 6.98 (t, J = 7.2 Hz, 2H, Ar-H), 7.08–7.23 (m, 10H, Ar-H) and 7.30–7.54 ppm (m, 8H, Ar-H); 13C-NMR (DMSO-d6): δ = 22.17, 29.70, 32.82 (CH2), 54.49 (pyrazole-CH), 81.43 (spiro-C), 120.93, 123.40, 127.10, 127.66, 128.17, 128.79, 129.24, 129.73, 129.97, 130.80, 132.07, 133.97, 134.39, 138.24, 138.92, 143.81, 146.38 and 206.09 ppm (Ar-C and CO); MS (EI): m/z (%) 800 (M+ + 2, 46.78), 799 (M+ + 1, 32.08), 798 (M+, 62.71); HRMS (EI): m/z calcd for C50H40Cl2N4O2 (M+) 798.2523, found 798.2523. Crystal data, C50H40Cl2N4O2, M = 799.76, triclinic, a = 11.4329(4) Å, b = 13.7041(5) Å, c = 15.7697(5) Å, V = 2244.11(13) Å3, α = 66.387(2)°, β = 82.782(2)°, γ = 89.224(2)°, space group: P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (#2), Z = 2, Dcalc = 1.292 g cm−3, no. of reflection measured 22943, 2 θmax = 66.66°, R1 = 0.0453.34
(#2), Z = 2, Dcalc = 1.292 g cm−3, no. of reflection measured 22943, 2 θmax = 66.66°, R1 = 0.0453.34
Bis-[1′-phenyl-4′-(4-nitrophenyl)-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] (4d). As yellow crystals, mp. 257–258 °C; IR (KBr): ν/cm−1 1683 (CO); 1H-NMR (DMSO-d6): δ = 1.05–1.10 (m, 2H, H of CH2), 1.41–1.45 (m, 2H, H of CH2), 1.94–1.98 (m, 2H, H of CH2), 2.27–2.32 (m, 2H, H of CH2), 2.66–2.70 (m, 2H, H of CH2), 2.84–2.89 (m, 2H, H of CH2), 5.07 (s, 2H, pyrazole-CH), 6.57 (d, J = 7.2 Hz, 2H, Ar-H), 6.71 (d, J = 7.8 Hz, 4H, Ar-H), 6.99 (t, J = 7.2 Hz, 2H, Ar-H), 7.12 (t, J = 7.8 Hz, 4H, Ar-H), 7.18 (d, J = 7.8 Hz, 4H, Ar-H), 7.38–7.42 (m, 6H, Ar-H) and 7.52 ppm (d, J = 8.4 Hz, 4H, Ar-H); 13C-NMR (DMSO-d6): δ = 21.36, 28.56, 31.14 (CH2), 58.03 (pyrazole-CH), 79.71 (spiro-C), 120.49, 122.94, 126.60, 127.12, 128.29, 128.33, 128.60, 131.20, 131.89, 132.15, 136.34, 138.02, 138.61, 143.44, 146.28 and 206.00 ppm (Ar-C and CO); MS (EI): m/z (%) 822 (M+ + 2, 9.89), 821 (M+ + 1, 27.65), 820 (M+, 45.12); HRMS (EI): m/z calcd for C50H40N6O6 (M+) 820.3004, found 820.3004. Crystal data, C50H40N6O6, M = 820.91, monoclinic, a = 13.204(5) Å, b = 9.770(4) Å, c = 16.496(6) Å, V = 2026(2) Å3, α = γ = 90°, β = 107.826(8)°, space group: P21/c (#14), Z = 4, Dcalc = 1.346 g cm−3, no. of reflection measured 15286, θmax = 50.10°, R1 = 0.0664.34
Bis-[1′-phenyl-4′-(4-anisyl)-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] (4e). As yellow crystals, mp. >300 °C; IR (KBr): ν/cm−1 1687 (CO); 1H-NMR (DMSO-d6): δ = 1.07–1.12 (m, 2H, H of CH2), 1.39–1.45 (m, 2H, H of CH2), 1.65–1.70 (m, 2H, H of CH2), 2.29–2.35 (m, 2H, H of CH2), 2.74–2.78 (m, 2H, H of CH2), 2.90–2.95 (m, 2H, H of CH2), 3.79 (s, 6H, 2OCH3), 4.91 (s, 2H, pyrazole-CH), 6.74 (d, J = 7.8 Hz, 4H, Ar-H), 6.86–6.90 (m, 10H, Ar-H), 7.14 (t, J = 7.8 Hz, 4H, Ar-H), 7.22 (d, J = 7.8 Hz, 2H, Ar-H), 7.27 (d, J = 7.8 Hz, 2H, Ar-H), 7.35 (t, J = 7.8 Hz, 2H, Ar-H) and 7.52 ppm (t, J = 7.8 Hz, 2H, Ar-H); 13C-NMR (CDCl3 at 50 °C): δ = 23.46, 29.08, 34.26 (CH2), 55.26 (OCH3) 59.12 (pyrazole-CH), 81.26 (spiro-C), 118.30, 121.16, 121.62, 126.60, 127.13, 128.34, 128.73, 129.09, 130.75, 131.90, 137.42, 139.39, 142.70, 144.07, 159.04 and 204.16 ppm (Ar-C and CO); MS (EI): m/z (%) 792 (M+ + 2, 20.97), 791 (M+ + 1, 60.25), 790 (M+, 100); HRMS (EI): m/z calcd for C52H46N4O4 (M+) 790.3513, found 790.3514.
Bis-[1′-phenyl-4′-(2,4-dimethoxyphenyl)-1-oxo-spiro-benzosub-erane-2,5′-pyrazoline] (4f). As yellow crystals, mp. >300 °C; IR (KBr): ν/cm−1 1685 (CO); 1H-NMR (DMSO-d6): δ = 1.29–1.34 (m, 2H, H of CH2), 1.48–1.53 (m, 2H, H of CH2), 1.62–1.65 (m, 2H, H of CH2), 2.30–2.35 (m, 2H, H of CH2), 2.78–2.86 (m, 2H, 4H of CH2), 3.69 (s, 6H, 2OCH3), 3.81 (s, 6H, 2OCH3), 5.20 (s, 2H, pyrazole-CH), 6.18 (d, J = 8.0 Hz, 2H, Ar-H), 6.45 (d, J = 8.0 Hz, 2H, Ar-H), 6.65–6.74 (m, 6H, Ar-H), 6.86 (t, J = 7.2 Hz, 2H, Ar-H), 7.08–7.15 (m, 4H, Ar-H), 7.22 (d, J = 7.6 Hz, 2H, Ar-H), 7.28 (d, J = 7.6 Hz, 2H, Ar-H), 7.37 (t, J = 7.6 Hz, 2H, Ar-H) and 7.51 ppm (t, J = 7.6 Hz, 2H, Ar-H); 13C-NMR (DMSO-d6): δ = 22.91, 29.34, 33.48 (CH2), 50.88 (pyrazole-CH), 55.85, 56.06 (OCH3), 81.25 (spiro-C), 98.91, 105.83, 117.17, 118.82, 121.64, 126.98, 128.64, 129.35, 129.49, 130.88, 132.10, 138.53, 139.81, 143.53, 144.98, 158.37, 160.82 and 205.43 ppm (Ar-C and CO); MS (EI): m/z (%) 852 (M+ + 2, 14.15), 851 (M+ + 1, 41.79), 850 (M+, 65.08); HRMS (EI): m/z calcd for C54H50N4O6 (M+) 850.3725, found 850.3725.
Bis-[1′-phenyl-4′-(4-tolyl)-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] (4g). As yellow crystals, mp. >300 °C; IR (KBr): ν/cm−1 1686 (CO); 1H-NMR (CDCl3 at 50 °C): δ = 1.31–1.35 (m, 2H, H of CH2), 1.48–1.52 (m, 2H, H of CH2), 1.78–1.81 (m, 2H, H of CH2), 2.31(s, 6H, 2CH3), 2.34–2.39 (m, 2H, H of CH2), 2.77–2.84 (m, 4H, 2H of CH2), 4.78 (s, 2H, pyrazole-CH), 6.60 (d, J = 7.2 Hz, 4H, Ar-H), 6.94 (d, J = 7.8 Hz, 4H, Ar-H), 7.14 (d, J = 7.8 Hz, 2H, Ar-H), 6.25–7.40 (m, 12H, Ar-H), 7.43 (t, J = 7.8 Hz, 2H, Ar-H) and 7.53 ppm (t, J = 7.8 Hz, 2H, Ar-H); 13C-NMR (CDCl3 at 50 °C): δ = 20.58 (CH3), 22.01, 29.15, 32.48 (CH2), 59.53 (pyrazole-CH), 81.27 (spiro-C), 118.90, 122.21, 126.53, 127.08, 127.45, 128.07, 128.84, 129.02, 129.73, 130.43, 131.76, 136.62, 137.70, 139.17, 143.71 and 204.78 ppm (Ar-C and CO); MS (EI): m/z (%) 760 (M+ + 2, 16.95), 759 (M+ + 1, 40.33), 758 (M+, 67.18); HRMS (EI): m/z calcd for C52H46N4O2 (M+) 758.3615, found 758.3614.
Bis-[1′-(4-chlorophenyl)-4′-phenyl-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] (4h). As yellow crystals, mp. >300 °C; IR (KBr): ν/cm−1 1687 (CO); 1H-NMR (CDCl3): δ = 1.32–1.36 (m, 2H, H of CH2), 1.50–1.55 (m, 2H, H of CH2), 1.77–1.82 (m, 2H, H of CH2), 2.32–2.39 (m, 2H, H of CH2), 2.80–2.82 (m, 4H, 2H of CH2), 4.75 (s, 2H, pyrazole-CH), 6.63 (d, J = 8.4 Hz, 4H, Ar-H), 7.07 (d, J = 8.4 Hz, 4H, Ar-H), 7.17 (d, J = 7.6 Hz, 2H, Ar-H), 7.31–7.38 (m, 8H, Ar-H), 7.43 (t, J = 7.6 Hz, 4H, Ar-H) and 757–7.61 ppm (m, 4H, Ar-H); 13C-NMR (CDCl3): δ = 23.34, 29.08, 34.23 (CH2), 59.81 (pyrazole-CH), 81.52 (spiro-C), 119.35, 126.29, 127.21, 127.64, 127.77, 128.26, 129.26, 129.51, 130.71, 132.16, 136.23, 137.06, 139.41, 141.14, 144.00 and 204.00 ppm (Ar-C and CO); MS (EI): m/z (%) 800 (M+ + 2, 41.25), 799 (M+ + 1, 31.08), 798 (M+, 49.98); HRMS (EI): m/z calcd for C50H40Cl2N4O2 (M+) 798.2523, found 798.2523.
Bis-[1′,4′-di-(4-chlorophenyl)-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] (4i). As yellow crystals, mp. 291–292 °C; IR (KBr): ν/cm−1 1682 (CO); 1H-NMR (DMSO-d6): δ = 1.06–1.08 (m, 2H, H of CH2), 1.43–1.45 (m, 2H, H of CH2), 1.96–1.98 (m, 2H, H of CH2), 2.26–2.30 (m, 2H, H of CH2), 2.69–2.71 (m, 2H, H of CH2), 2.84–2.89 (m, 2H, H of CH2), 5.03 (s, 2H, pyrazole-CH), 6.72 (d, J = 8.4 Hz, 4H, Ar-H), 7.15 (d, J = 8.4 Hz, 4H, Ar-H), 7.19–7.25 (m, 4H, Ar-H), 7.31–7.41 (m, 6H, Ar-H), 7.44 (t, J = 7.8 Hz, 2H, Ar-H) and 7.50 ppm (d, J = 8.4 Hz, 4H, Ar-H); 13C-NMR (DMSO-d6): δ = 21.46, 28.55, 31.37 (CH2), 58.06 (pyrazole-CH), 80.08 (spiro-C), 119.52, 121.38, 126.53, 126.77, 127.53, 128.18, 128.39, 128.87, 132.21, 132.27, 135.99, 138.17, 138.36, 142.35, 146.24 and 205.25 ppm (Ar-C and CO); MS (EI): m/z (%) 868 (M+ + 2, 52.28), 867 (M+ + 1, 22.05), 866 (M+, 37.48); HRMS (EI): m/z calcd for C50H38Cl4N4O2 (M+) 866.1743, found 866.1743.
Bis-[1′-(4-chlorophenyl)-4′-(2,4-dimethoxyphenyl)-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] (4j). As yellow crystals, mp. >300 °C; IR (KBr): ν/cm−1 1685 (CO); 1H-NMR (CDCl3): δ = 1.55–1.58 (m, 2H, H of CH2), 1.76–1.80 (m, 2H, H of CH2), 2.34–1.41 (m, 2H, H of CH2), 2.76–2.81 (m, 2H, H of CH2), 2.87–2.91 (m, 2H, H of CH2), 3.09–3.13 (m, 2H, H of CH2), 3.63 (s, 6H, 2OCH3), 3.86 (s, 6H, 2OCH3), 5.31 (s, 2H, pyrazole-CH), 6.27 (d, J = 8.4 Hz, 2H, Ar-H), 6.34 (d, J = 8.4 Hz, 2H, Ar-H), 6.47 (s, 2H, Ar-H), 6.68 (d, J = 7.6 Hz, 4H, Ar-H), 7.05 (d, J = 8.0 Hz, 4H, Ar-H), 7.14 (d, J = 7.6 Hz, 2H, Ar-H), 7.33 (t, J = 7.6 Hz, 2H, Ar-H), 7.44 (t, J = 7.6 Hz, 2H, Ar-H) and 7.52 ppm (d, J = 7.6 Hz, 2H, Ar-H); 13C-NMR (CDCl3): δ = 23.60, 29.66, 34.34 (CH2), 50.91 (pyrazole-CH), 55.56, 55.58 (OCH3), 81.47 (spiro-C), 97.85, 104.67, 117.61, 119.14, 127.15, 127.82, 128.33, 129.02, 129.37, 130.84, 131.44, 131.86, 138.01, 140.03, 144.97, 158.16, 160.54 and 204.74 ppm (Ar-C and CO); MS (EI): m/z (%) 920 (M+ + 2, 55.82), 919 (M+ + 1, 41.98), 918 (M+, 66.13), 887 (40.07), 799 (100), 459 (M+/2, 9.25); HRMS (EI): m/z calcd for C54H48Cl2N4O6 (M+) 918.2945, found 918.2946.
Bis-[1′-(4-tolyl)-4′-phenyl-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] (4k). As yellow crystals, mp. 274–275 °C; IR (KBr): ν/cm−1 1687 (CO); 1H-NMR (DMSO-d6): δ = 0.97–1.01 (m, 2H, H of CH2), 1.32–1.35 (m, 2H, H of CH2), 1.59–1.62 (m, 2H, H of CH2), 2.17 (s, 6H, 2CH3), 2.24–2.28 (m, 2H, H of CH2), 2.68–2.72 (m, 2H, H of CH2), 2.86–2.92 (m, 2H, H of CH2), 4.93 (s, 2H, pyrazole-CH), 6.59 (d, J = 8.4 Hz, 4H, Ar-H), 6.84–6.88 (m, 4H, Ar-H), 6.92 (d, J = 8.4 Hz, 4H, Ar-H), 7.17 (d, J = 7.6 Hz, 2H, Ar-H), 7.23 (d, J = 7.6 Hz, 2H, Ar-H), 7.29–7.33 (m, 8H, Ar-H) and 7.40 ppm (t, J = 7.6 Hz, 2H, Ar-H); 13C-NMR (DMSO-d6): δ = 20.18 (CH3), 21.65, 28.06, 31.88 (CH2), 58.76 (pyrazole-CH), 79.32 (spiro-C), 118.86, 120.80, 126.82, 128.08, 128.41, 128.85, 129.11, 129.17, 130.82, 132.23, 136.47, 137.94, 138.91, 140.26, 144.00 and 205.64 ppm (Ar-C and CO); MS (EI): m/z (%) 760 (M+ + 2, 8.55), 759 (M+ + 1, 30.29), 758 (M+, 56.11), 639 (100), 379 (M+/2, 6.77); HRMS (EI): m/z calcd for C52H46N4O2 (M+) 758.3615, found 758.3614.
Bis-[1′-(4-tolyl)-4′-(4-chlorophenyl)-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] (4l). As yellow crystals, mp. >300 °C; IR (KBr): ν/cm−1 1686 (CO); 1H-NMR (CDCl3): δ = 1.30–1.35 (m, 2H, H of CH2), 1.53–1.57 (m, 2H, H of CH2), 1.74–1.79 (m, 2H, H of CH2), 2.21–2.24 (m, 2H, H of CH2), 2.26 (s, 6H, 2CH3), 2.36–2.43 (m, 2H, H of CH2), 2.78–2.80 (m, 4H, H of CH2), 4.74 (s, 2H, pyrazole-CH), 6.65–6.81 (m, 8H, Ar-H), 6.97 (d, J = 8.0 Hz, 4H, Ar-H), 7.14 (d, J = 7.6 Hz, 2H, Ar-H), 7.24 (d, J = 8.0 Hz, 4H, Ar-H), 7.32 (t, J = 7.6 Hz, 2H, Ar-H) and 7.41–7.47 ppm (m, 4H, Ar-H); 13C-NMR (CDCl3): δ = 20.82 (CH3), 23.40, 29.32, 34.20 (CH2), 58.83 (pyrazole-CH), 81.50 (spiro-C), 119.40, 122.55, 127.35, 128.58, 129.21, 130.37, 131.24, 131.78, 132.10, 133.68, 135.47, 137.87, 139.05, 140.42, 143.71 and 204.69 ppm (Ar-C and CO); MS (EI): m/z (%) 828 (M+ + 2, 50.78), 827 (M+ + 1, 36.08), 826 (M+, 60.25), 707 (100), 413 (M+/2, 5.23); HRMS (EI): m/z calcd for C52H44Cl2N4O2 (M+) 826.2836, found 826.2835. Crystal data, C52H44Cl2N4O2, M = 827.86, monoclinic, a = 13.155(2) Å, b = 14.042(2) Å, c = 13.208(2) Å, V = 2124.1(6) Å3, α = γ = 90°, β = 119.470(9)°, space group: P21/c (#14), Z = 4, Dcalc = 1.294 g cm−3, no. of reflection measured 4836, θmax = 54.9°, R1 = 0.0467.34
Bis-[1′-(4-tolyl)-4′-(2,4-dimethoxyphenyl)-1-oxo-spirobenzosub-erane-2,5′-pyrazol-ine] (4m). As yellow crystals, mp. >300 °C; IR (KBr): ν/cm−1 1686 (CO); 1H-NMR (CDCl3): δ = 1.38–1.42 (m, 2H, H of CH2), 1.51–1.54 (m, 2H, H of CH2), 1.74–1.76 (m, 2H, H of CH2), 2.23 (s, 6H, 2CH3), 2.32–2.36 (m, 2H, H of CH2), 2.75–2.78 (m, 2H, H of CH2), 2.89–2.91 (m, 2H, H of CH2), 3.65 (s, 6H, 2OCH3), 3.85 (s, 6H, 2OCH3), 5.37 (s, 2H, pyrazole-CH), 6.31 (t, J = 8.4 Hz, 2H, Ar-H), 6.48 (s, 2H, Ar-H), 6.68 (d, J = 7.8 Hz, 4H, Ar-H), 6.91 (d, J = 7.8 Hz, 4H, Ar-H), 7.12 (d, J = 7.8 Hz, 2H, Ar-H), 7.32 (t, J = 8.4 Hz, 2H, Ar-H), 7.38–7.41 (m, 4H, Ar-H) and 7.49 ppm (d, J = 8.4 Hz, 2H, Ar-H); 13C-NMR (CDCl3): δ = 20.55 (CH3), 23.30, 29.47, 33.99 (CH2), 50.42 (pyrazole-CH), 55.31, 55.53 (2OCH3) 80.99 (spiro-C), 97.78, 104.43, 118.37, 123.28, 126.29, 126.76, 128.57, 129.31, 129.76, 130.33, 131.27, 131.38, 138.37, 139.58, 144.37, 158.05, 160.10 and 205.30 ppm (Ar-C and CO); MS (EI): m/z (%) 880 (M+ + 2, 11.29), 879 (M+ + 1, 34.05), 878 (M+, 53.97), 759 (100), 439 (M+/2, 7.31), 405 (57.11); HRMS (EI): m/z calcd for C56H54N4O6 (M+) 878.4038, found 878.4038.
Conclusions
A direct and efficient one-pot 1,3-dipolar cycloaddition reaction of bis-hydrazonoyl chlorides 1a–c with 2-arylidene-1-benzosuberone derivatives 3a–g to give a series of novel class of the bis-[1′,4′-diaryl-1-oxo-spiro-benzosuberane-2,5′-pyrazoline] derivatives 4a–m has been developed. Ultrasonic irradiation proved to be a superior and an efficient tool for promotion of such 1,3-dipolar cycloaddition reactions using ethanol as solvent and Et3N as a base. The cycloaddition was elucidated as regio- and stereoselective process as examined by measuring the X-ray crystallographic data of five examples of the obtained cycloadducts.
Acknowledgements
The financial support for this research work was provided by the University of Kuwait through a research grant (SC03/14). The facilities of ANALAB and SAF supported by research grants GS01/01, GS01/05, GS01/03, and GS03/08 are gratefully acknowledged.
Notes and references
- T. J. Mason and J. P. Lorimer, Sonochemistry: Theory, Applications and Uses of Ultrasound in Chemistry, Ellis Horwood Ltd., 1988 Search PubMed.
- T. J. Mason and D. Peters, Practical Sonochemistry, Ellis Horwood, New York, NY, USA, 1991 Search PubMed.
- J. L. Luche, Organic Sonochemistry, Plenum Press, New York, NY, USA, 1998 Search PubMed.
- T. J. Mason and J. P. Lorimer, Applied sonochemistry: uses of power ultrasound in chemistry and processing, Wiley-VCH, Weinheim, 2002 Search PubMed.
- G. Cravotto and P. Cintas, Chem. Soc. Rev., 2006, 35, 180–196 RSC.
- A. Bazgir, S. Ahadi, R. Ghahremanzadeh, H. R. Khavasi and P. Mirzaei, Ultrason. Sonochem., 2010, 17, 447–452 CrossRef CAS PubMed.
- J. L. G. Ruano, A. Parra, L. Marzo, F. Yuste and V. M. Mastranzo, Tetrahedron, 2011, 67, 2905–2910 CrossRef PubMed.
- A. Padwa and W. H. Pearson, Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, Wiley-Interscience, 1st edn, 2002 Search PubMed.
- A. Padwa, Intramolecular 1,3-Dipolar Cycloaddition, in Comprehensive Organic Chemistry, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 4, pp. 1069 Search PubMed.
- W. L. Chan, M. Ding and B. Zou, PCT Int. Appl, WO 2014167528 A1 20141016, 2014.
- K. M. Dawood, J. Heterocycl. Chem., 2005, 42, 221–225 CrossRef CAS.
- Â. Monteiro, L. M. Gonçalves and M. M. M. Santos, Eur. J. Med. Chem., 2014, 79, 266–272 CrossRef PubMed.
- M. Kamata and T. Yamashita, Jpn. Kokai Tokkyo Koho, JP 2009196966 A 20090903, 2009.
- L. Nagarapu, B. Yadagiri, R. Bantu, C. G. Kumar, S. Pombala and J. Nanubolu, Eur. J. Med. Chem., 2014, 71, 91–97 CrossRef CAS PubMed.
- C. Schmitt, M. Voegelin, A. Marin, M. Schmitt, F. Schegg, P. Henon, D. Guenot and C. Tarnus, Bioorg. Med. Chem., 2013, 21, 2135–2144 CrossRef CAS PubMed.
- R. P. Tanpure, C. S. George, M. Sriram, T. E. Strecker, J. K. Tidmore, E. Hamel, A. K. Charlton-Sevcik, D. J. Chaplin, M. L. Trawick and K. G. Pinney, MedChemComm, 2012, 3, 720–724 RSC.
- B. Yadagiri, U. D. Holagunda, R. Bantu, L. Nagarapu, V. Guguloth, S. Polepally and N. Jain, Bioorg. Med. Chem. Lett., 2014, 24, 5041–5044 CrossRef CAS PubMed.
- S. Bhat, J. S. Shim and J. O. Liu, Bioorg. Med. Chem. Lett., 2013, 23, 2733–2737 CrossRef CAS PubMed.
- M. Pilatova, L. Varinska, P. Perjesi, M. Sarissky, L. Mirossay, P. Solar, A. Ostro and J. Mojzis, Toxicol. In Vitro, 2010, 24, 1347–1355 CrossRef CAS PubMed.
- C. Maiereanu, C. Schmitt, N. Schifano-Faux, D. Le Nouen, A. Defoin and C. Tarnus, Bioorg. Med. Chem., 2011, 19, 5716–5733 CrossRef CAS PubMed.
- S. Albrecht, M. Al-Lakkis-Wehbe, A. Orsini, A. Defoin, P. Pale, E. Salomon, C. Tarnus and J.-M. Weibel, Bioorg. Med. Chem., 2011, 19, 1434–1449 CrossRef CAS PubMed.
- J.-M. Contreras, I. Parrot, W. Sippl, Y. M. Rival and C. G. Wermuth, J. Med. Chem., 2001, 44, 2707–2718 CrossRef CAS PubMed.
- K. M. Dawood, Tetrahedron, 2005, 61, 5229–5233 CrossRef CAS PubMed.
- K. M. Dawood and N. M. Elwan, J. Chem. Res., 2004, 4, 264–266 CrossRef.
- K. M. Dawood, M. A. Raslan and A. M. Farag, Synth. Commun., 2003, 33, 4075–4086 CrossRef PubMed.
- A. S. Shawali, A. M. Farag, M. S. Algharib and K. M. Dawood, Tetrahedron, 1994, 50, 5091–5098 CrossRef.
- A. M. Farag, Z. E. Kandeel, M. S. Algharib and K. M. Dawood, Phosphorus, Sulfur Silicon Relat. Elem., 1994, 91, 129–136 CrossRef CAS.
- A. S. Shawali, A. M. Farag, H. A. Albar and K. M. Dawood, Tetrahedron, 1993, 49, 2761–2766 CrossRef CAS.
- A. M. Farag, A. S. Shawali, N. M. Abed and K. M. Dawood, Gazz. Chim. Ital., 1993, 123, 467–470 CAS.
- H. Behbehani, H. M. Ibrahim and M. H. Elnagdi, Tetrahedron, 2013, 69, 6176–6184 CrossRef CAS PubMed.
- H. Behbehani and H. M. Ibrahim, Tetrahedron, 2013, 69, 10535–10543 CrossRef CAS PubMed.
- R. Raghunathan, M. Shanmugasundaram, S. Bhanumathi and P. J. E. Malar, Heteroat. Chem., 1998, 9, 327–332 CrossRef CAS.
- H. M. Hassaneen, R. H. Hilal, N. M. Elwan, A. Harhash and A. S. Shawali, J. Heterocycl. Chem., 1984, 21, 1013–1016 CrossRef CAS.
- ESI.†.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112–122 CrossRef PubMed.
- N. R. El-Rayyes and N. H. Bahtiti, J. Heterocycl. Chem., 1989, 26, 209–214 CrossRef CAS.
- C. Grundmann, S. K. Datta and R. F. Sprecher, Liebigs Ann. Chem., 1971, 744, 88–104 CrossRef CAS.
- K. M. Dawood, Synthesis and reactivity of some nitrogen and phosphorus dipoles, M.Sc. thesis, Cairo University, 1992.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.