
One-pot highly enantio- and diastereoselective synthesis of anti,anti vinylic 3-amino-1,2 diols via proline catalyzed sequential α-amination/ benzoyloxyallylation of aldehydes†
Brij Bhushan Ahuja and
Arumugam Sudalai*
Chemical Engineering & Process Development Division, National Chemical Laboratory, Dr Homi Bhabha Road, Pune-411008, Maharashtra, India. E-mail: a.sudalai@ncl.res.in
First published on 24th February 2015
Abstract
The first direct asymmetric synthesis of anti,anti vinylic 3-amino-1,2-diols from aldehydes is described via a one-pot sequential L-proline catalyzed α-amination/benzoyloxyallylation protocol. The reaction proceeds with exceptionally high diastereoselectivity (>99%) as can be explained based on the Felkin–Ahn transition state model. Its effectiveness is proven unambiguously by demonstrating a short asymmetric synthesis of D-ribo-phytosphingosine tetraacetate (93% ee).
Vicinal amino diol subunits with three contiguous, heteroatom-bearing chiral carbons, constitute an important stereotriad pattern found in numerous pharmaceuticals and bioactive natural products.1 The majority of synthetic strategies for these motifs often employ starting materials from the chiral pool2a,b or utilize the ring-opening of chiral epoxy alcohols with amine nucleophiles.2c A number of diastereoselective syntheses of these molecules have also been reported; some recent examples include the addition of Grignard reagents to α-substituted nitriles,2d imines2e or catalytic processes involving dihydroxylation of allylamines,2f pinnacol coupling of chiral α-amino aldehydes2g and an oxidation/reduction sequence of bicyclic methyleneaziridines.2h However, the efficient construction of vicinal amino diol units with well-defined stereochemistry and derivatizable functional groups remains a challenge in organic synthesis. To the best of our knowledge, a direct method of synthesis for these stereotriads utilizing aldehydes as precursor remains elusive.
The synthetic methodology leading rapidly to structural complexity from readily available starting material through one-pot reaction sequence is widely recognized.3 In particular, proline catalyzed sequential reaction such as α-amination of aldehydes4 followed by Wittig,5a,b aldol,5c or Corey–Chaykovsky5d have gained more prominence in recent years. This is necessitated because α-aminated aldehyde is prone to racemisation during isolation. In this communication, we wish to describe a one-pot procedure for a tandem α-amination/benzoyloxyallylation of aldehydes 1a–j that proceeds to give vicinal amino diols 2a–j in a highly enantio- and diastereoselective fashion (Scheme 1).
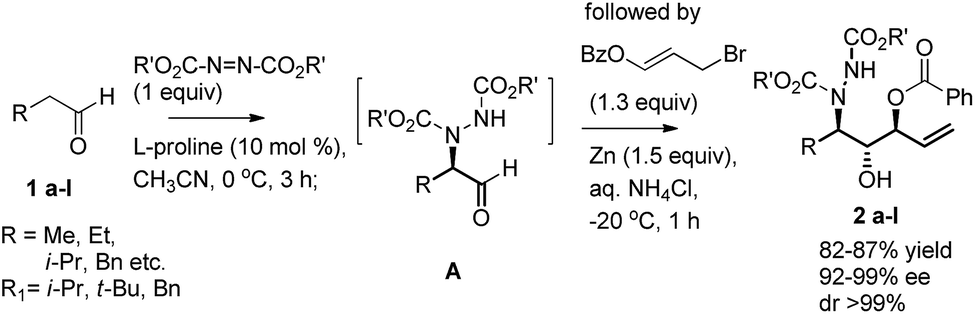 | ||
| Scheme 1 In situ trapping of α-amino aldehydes (A) with benzoyloxyallyl bromide. | ||
In the initial study, propanal 1a was α-aminated with diisopropyl azodicarboxylate (DIAD) catalyzed by L-proline (10 mol%) CH3CN at 0 °C for 3 h that produced the corresponding α-aminated aldehyde (A) in situ, followed by the sequential addition of Zn powder (1.5 equiv.), 3-benzoyloxyallyl bromide6 (1.5 equiv.) and saturated aq. NH4Cl at 0 °C, gave anti,anti vinylic 3-amino-1,2-diol 2a in 79% yield (dr = 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). Also, its diastereoselectivity could be marginally improved to 4:1 when the reaction was conducted at −10 °C. Finally, at −20 °C, we observed that 2a could be obtained as a single diastereomer with dr 99:1 and 77% ee. The subsequent investigation has shown that dibenzyl azadicarboxylate was found to be an excellent amine sources for this sequential reaction (entry 5) (Table 1).
:3). Also, its diastereoselectivity could be marginally improved to 4:1 when the reaction was conducted at −10 °C. Finally, at −20 °C, we observed that 2a could be obtained as a single diastereomer with dr 99:1 and 77% ee. The subsequent investigation has shown that dibenzyl azadicarboxylate was found to be an excellent amine sources for this sequential reaction (entry 5) (Table 1).
|
|||||
|---|---|---|---|---|---|
| No. | R′ | T (°C) | Product (2a) | ||
| Yieldb (%) | eec (%) | drc | |||
a Propanaldehyde (5 mmol), amine (R′O2C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–CO2−R′) (5 mmol), L-proline (10 mol%), 3-benzoyloxyallyl bromide (7.5 mmol), Zn (7.5 mmol), saturated aq. NH4Cl (10 mL).b Isolated yield.c From chiral HPLC analysis. N–CO2−R′) (5 mmol), L-proline (10 mol%), 3-benzoyloxyallyl bromide (7.5 mmol), Zn (7.5 mmol), saturated aq. NH4Cl (10 mL).b Isolated yield.c From chiral HPLC analysis. |
|||||
| 1 | iPr | 0 | 79 | 69 | 7:3 |
| 2 | iPr | −10 | 79 | 77 | 4:1 |
| 3 | iPr | −20 | 79 | 77 | 99:1 |
| 4 | tBu | −20 | 81 | 78 | 99:1 |
| 5 | Bn | −20 | 84 | 93 | 99:1 |

To extend the scope of this one-pot reaction, a series of aliphatic aldehydes bearing different functionalities (alkyl, aryl, alkenyl, benzoyloxy or methoxy methyl) were examined under the optimized condition (Table 2). For all the cases studied, the products 2a–j were indeed obtained in high yields (81–87%) and excellent enantioselectivity (91–99%) with de > 99%. The stereochemical assignment of this sequential reaction was made based on previously established absolute configuration of α-amino aldehydes.4a The anti,anti stereochemistry in 2a was proven unambiguously from X-ray crystallographic analysis (Fig. 1).
| No. | Aldehydes (R) (1a–j) | Products (2a–j) | |||
|---|---|---|---|---|---|
| Amines (R′) | Yieldb (%) | eec (%) | dec (%) | ||
| a Aldehyde (5 mmol), amine (R′O2C–NN–CO2−R′) (5 mmol), L-proline (10 mol%), CH3CN (25 mL), 0 °C, 3 h followed by 3-benzoyloxyallyl bromide (7.5 mmol), Zn (7.5 mmol), saturated aq. NH4Cl (10 mL), −20 °C, 2 h.b Isolated yield.c From chiral HPLC analysis. |
|||||
| 1 | Methyl (1a) | Bn | 84 | 93 | >99 |
| 2 | Ethyl (1b) | iPr | 87 | 91 | >99 |
| 3 | i-Propyl (1c) | iPr | 83 | 95 | >99 |
| 4 | n-Propyl (1d) | iPr | 86 | 93 | >99 |
| 5 | 3-(Methoxymethoxy)-ethyl (1e) | iPr | 87 | 93 | 99 |
| 6 | 3-(Benzyloxy)ethyl (1f) | tBu | 84 | 93 | 99 |
| 7 | But-3-enyl (1g) | iPr | 82 | 95 | >99 |
| 8 | Benzyl (1h) | tBu | 84 | 97 | >99 |
| 9 | 4-Methoxybenzyl (1i) | tBu | 81 | 99 | >99 |
| 10 | 2-(Benzyloxy)methyl (1j) | Bn | 85 | 93 | >99 |
 | ||
| Fig. 1 ORTEP diagram of compound 2a. | ||
To rationalize the observed high anti,anti diastereoselectivity of vicinal amino diols, both Felkin–Ahn6d (TS 1) and six membered transition state (TS II) model have been proposed (Fig. 2). Anti relationship at C1–C2 carbons is governed by the Felkin–Ahn model in which Zn atom of benzoyloxyallylzinc reagent is coordinated to the carbonyl oxygen and the nucleophilic attack of the corresponding reagent takes place at ‘Si’ face predominantly perpendicular to the bulky R1N–NHR1 group. Also, anti relationship at C2–C3 carbons can be explained based on the six membered transition state model (TS II) in which hydrazino alkyl group of aldehyde and OBz group of nucleophile are oriented in the pseudoequatorial position to deliver anti diol.
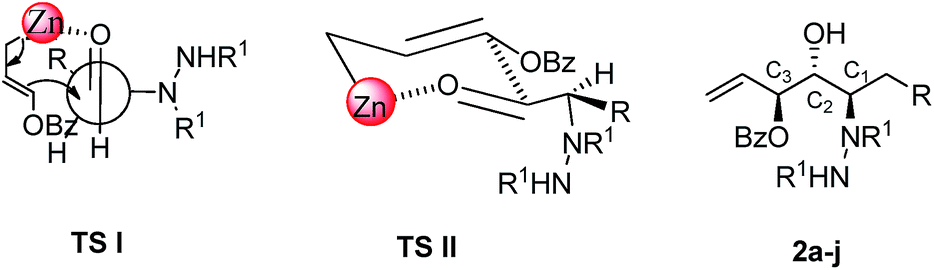 | ||
| Fig. 2 Proposed transition state model (R1 = CO2iPr, CO2tBu, CO2Bn; R = alkyl, alkyl aryl). | ||
To further extend its synthetic utility 1a was subjected to hydroboration/oxidation sequence that gave functionalized amino triol 3 in high yield. Also 1c was hydrogenated over RANEY® Ni followed by its Boc protection giving 4 in 89% yield (Scheme 2).
 | ||
| Scheme 2 Oxidative and reductive transformations of anti,anti vinylic 3-amino-1,2-diols. | ||
Finally, a short enantioselective synthesis of D-ribo-phytosphingosine tetraacetate7 (7) seemed attractive to us because it is a bioactive lipid that has potential antitumor properties8 (Scheme 3). Its synthesis was achieved in 5 steps commencing from aldehyde 1j, which was subjected to D-proline catalyzed sequential α-amination/benzoyloxyallylation protocol to afford vinylic aminodiol ent-2j (85%, 93% ee). The LiOH-mediated hydrolysis of ent-2j gave oxazolidinone 5 in 75% yield. The cross-metathesis of 5 with 1-tetradecene over Grubbs' catalyst produced 6 (72% yield). The catalytic hydrogenation [RANEY® Ni, H2 (60 psig), 24 h] of 6 followed by basic hydrolysis (K2CO3, MeOH) and its acetylation (Ac2O, py, DMAP) produced the target phytosphingosine 7 in 76% yield and 93% ee.
 | ||
| Scheme 3 Synthesis of D-ribo-phytosphingosine tetraacetate (7). | ||
In conclusion, we have described an unprecedented, one-pot procedure for a sequential α-amination/benzoyloxyallylation of aldehydes that leads to the synthesis of vinylic-3-amino-1,2-diols 2a–l in high yields with excellent enantio- and diastereoselectivities. This protocol generates three chiral centers consecutively with anti,anti relationship in a single step, and has been successfully applied to the short asymmetric synthesis of D-ribo-phytosphingosine tetraacetate, 7. We believe this one-pot sequential method will find tremendous application in the synthesis of bioactive natural products and pharmaceutical substances.
Acknowledgements
BBA thanks CSIR, New Delhi for the award of Senior Research Fellowship and DST (no. SR/S1/OC-67/2010), New Delhi for financial support. Authors thank Dr V. V. Ranade, Chair and Head, Chemical Engineering and Process Development Division for his constant encouragement.Notes and references
- (a) D. H. Rich, in Comprehensive Medicinal Chemistry, ed. P. G. Sammes, Pergamon Press, Oxford, 1990, vol. 2, p. 391 Search PubMed; (b) S. C. Bergmeier, Tetrahedron, 2000, 56, 2561 CrossRef CAS.
- (a) S. Umezawa, Pure Appl. Chem., 1978, 50, 1453 CrossRef CAS; (b) X. Liang, J. Andersch and M. Bols, J. Chem. Soc., Perkin Trans. 1, 2001, 2136 RSC; (c) C. Wang and H. Yamamoto, J. Am. Chem. Soc., 2014, 136, 6888 CrossRef CAS PubMed; (d) P. Hutin and M. Larcheveque, Tetrahedron Lett., 2000, 41, 2369 CrossRef CAS; (e) N. Meunier, U. Veith and V. Jager, Chem. Commun., 1996, 331 RSC; (f) J. S. Oh, J. Jeon, D. Y. Park and Y. G. Kim, Chem. Commun., 2005, 770 RSC; (g) A. W. Konradi and S. F. Pedersen, J. Org. Chem., 1990, 55, 4506 CrossRef CAS; (h) J. W. Rigoli, I. A. Guzei and J. M. Schomaker, Org. Lett., 2014, 16, 1696 CrossRef CAS PubMed , and references cited therein.
- (a) H. Sundén, I. Ibrahem, L. Eriksson and A. Córdova, Angew. Chem., Int. Ed., 2005, 44, 4877 CrossRef PubMed; (b) B. List, J. Am. Chem. Soc., 2000, 122, 9336 CrossRef CAS; (c) D. B. Ramachary, Y. V. Reddy and M. Kishor, Org. Biomol. Chem., 2008, 6, 4188 RSC; (d) D. B. Ramachary, C. Venkaiah and Y. V. Reddy, Org. Biomol. Chem., 2014, 12, 5400 RSC; (e) M. Gruttadauria, L. A. Bivona, P. L. Meo, S. Riela and R. Noto, Eur. J. Org. Chem., 2012, 2635 CrossRef CAS.
- (a) B. List, J. Am. Chem. Soc., 2002, 124, 5656 CrossRef CAS PubMed; (b) A. Bogevig, K. Juhl, N. Kumaragurubaran, W. Zhuang and K. A. Jorgensen, Angew. Chem., Int. Ed., 2002, 41, 1790 CrossRef CAS; (c) N. Kumaragurubaran, K. Juhl, W. Zhuang, A. Bogevig and K. A. Jorgensen, J. Am. Chem. Soc., 2002, 124, 6254 CrossRef CAS PubMed; (d) H. Vogt, S. Vanderheiden and S. Brase, Chem. Commun., 2003, 2448 RSC , and references cited therein.
- (a) A. J. Oelke, S. Kumarn, D. A. Longbottom and S. V. Ley, Synlett, 2006, 2548 CAS; (b) S. P. Kotkar, V. B. Chavan and A. Sudalai, Org. Lett., 2007, 9, 1001 CrossRef CAS PubMed; (c) N. S. Chowdari, D. B. Ramachary and C. F. Barbas, Org. Lett., 2003, 5, 1685 CrossRef CAS PubMed; (d) B. S. Kumar, V. Venkataramasubramanian and A. Sudalai, Org. Lett., 2012, 14, 2468 CrossRef CAS PubMed; (e) V. Rawat, B. S. Kumar and A. Sudalai, Org. Biomol. Chem., 2013, 11, 3608 RSC.
- (a) L. Keinicke, P. Fristrup, P. O. Norrby and R. Madsen, J. Am. Chem. Soc., 2005, 127, 15756 CrossRef CAS PubMed; (b) M. Lombardo, R. Girotti, S. Morganti and C. Trombini, Chem. Commun., 2001, 2310 RSC; (c) M. Lombardo, R. Girotti, S. Morganti and C. Trombini, Org. Lett., 2001, 3, 2981 CrossRef CAS PubMed; (d) M. Liu, X. W. Sun, M. H. Xu and G. Q. Lin, Chem.–Eur. J., 2009, 15, 10217 CrossRef CAS PubMed , and references cited therein.
- (a) M. Miroslava, P. Kvetoslava and G. Jozef, Chem. Pap., 2013, 67, 84 Search PubMed; (b) R. Mettu, N. R. Thatikonda, O. S. Olusegun, R. Vishvakarma and J. R. Vaidya, ARKIVOC, 2012, 421 CrossRef CAS; (c) G. S. Rao and B. V. Rao, Tetrahedron Lett., 2011, 52, 4861 CrossRef PubMed.
- (a) E. Kobayashi, K. Motoski, Y. Yamaguchi, T. Uchida, H. Fukushima and Y. Koezuka, Oncol. Res., 1995, 7, 529 CAS; (b) R. D. Goff, Y. Gao, J. Mattner, D. Zhou, N. Yin, C. Cantu, L. Teyton, A. Bendelac and P. B. Savage, J. Am. Chem. Soc., 2004, 126, 13602 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, IR, and HRMS of new compounds. CCDC 1026336. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra02830b |
| This journal is © The Royal Society of Chemistry 2015 |