DOI:
10.1039/C5RA02680F
(Paper)
RSC Adv., 2015,
5, 34125-34133
Influence of calcination temperature on the catalytic performance of Co3O4/GO nanocomposites for Orange II degradation†
Received
12th February 2015
, Accepted 7th April 2015
First published on 7th April 2015
Abstract
In this study, graphene oxide-supported cobalt oxide catalysts (Co3O4/GO), which are used to degrade Orange II from water via advanced oxidation processes based on sulfate radicals, are synthesized in situ as heterogeneous catalysts via a solvothermal method. The catalysts were calcined at different temperatures of 300 °C, 500 °C, 700 °C, and 900 °C for 2 h in N2 atmosphere. The catalytic activities of both uncalcined and calcined catalysts were studied at the same time. The results show that all the formed catalysts exhibit high catalytic activity, and the catalysts calcined at 900 °C have the best activity for the degradation of Orange II. X-ray diffraction (XRD), thermal gravimetric analysis (TGA), Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), and transmission electron microscopy (TEM) are employed to study the mechanism behind the change in catalytic activity. The analysis indicates that the change in the quantity of hydroxylated cobalt (Co–OH), the valence state of cobalt oxide and the reduction degree of graphene oxide (GO), which are all caused by calcination, are responsible for the change in catalytic activity.
1. Introduction
Advanced oxidation processes based on sulfate radicals (AOPs-SRs) have gained extensive attention over the past years as a promising technique to completely degrade most organic pollutants.1 Thus far, peroxymonosulfate (PMS) coupled with Co2+ has been proven to be the best combination for the generation of sulfate radicals (SO4−˙) for Orange II degradation.2 However, the degradation reaction in a homogeneous system has several disadvantages because of cobalt leachate, which has biological toxicity and is recognized as a priority metal pollutant.3 Moreover, the Co2+ as catalysts cannot be reused. Therefore, great attention has been recently given in the activation of PMS by heterogeneous cobalt sources.4 Our previous studies5 found that cobalt oxide catalyst immobilized on graphene oxide (Co3O4/GO) was successfully synthesized as a heterogeneous catalyst for PMS activation to generate SO4−˙. Meanwhile, the experiments showed that Co3O4/GO composites function more efficiently for PMS activation than either pure Co3O4 or bare GO alone. These phenomena suggested a synergetic catalytic effect of Co3O4 and GO in the hybrid state. Whether hydroxylated cobalt (Co–OH) formed on the surface of the GO sheet via direct interaction of Co species with nearby hydroxyl groups or through the dissociation of H2O with Co2+ is a critical impact factor for radical generation in heterogeneous activation of PMS.6
The catalytic activity of catalysts is influenced by factors such as calcination temperature, preparation procedure, nature of support, doping with foreign ions, and the active phase precursor employed.7 Specifically, the calcination temperatures typically affect the morphology and chemical properties of the catalysts. Co3O4 belongs to the family of complex-metal oxides known for their spinel structures that consist of Co2+ cations in tetrahedral sites and Co3+ cations in octahedral sites. As such, two kinds of oxygen ions exist, where one is bonded to three Co3+ ions and the other is bonded to one Co3+ and one Co2+ ion as the nearest neighbors.8,9 In this structure, accordingly, two species of cobalt oxides exist, which are CoO and Co3O4. Both are stable in the natural environment.10,11 The catalytic activity of Co3O4 is related to the ratio of Co3+ and Co2+ on the predominantly exposed planes.12 However, the morphology and chemical properties of the catalysts heavily depend on the calcination temperature. High temperature calcination may greatly change the special structure and surface redox reactivity of Co3O4. The graphite oxide support is a solid with a C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O ratio between 2.1 and 2.9 and has multiple oxygen-containing functional groups, such as hydroxyls and epoxides in the basal plane, and carboxyl groups at plane edges.13 Graphite oxide consists of loosely bound layers. Each layer has a two-dimensional arrangement of carbon atoms in the graphene pattern with a thickness of approximately 1.1 nm.14 The oxygen-containing functional groups can easily fall off from the surface of graphite oxide through calcination in inert gas atmosphere because of its special structure. Meanwhile, graphite oxide is exfoliated and turns into graphene oxide, reduced graphene oxide (rGO), or graphene based on different calcination temperatures. The present heterogeneous catalysts Co3O4/GO are fabricated through supporting cobalt oxide on the surface of graphene oxide. Calcination may significantly influence the catalytic performance of Co3O4/GO, but the correlated research is insufficient.
:O ratio between 2.1 and 2.9 and has multiple oxygen-containing functional groups, such as hydroxyls and epoxides in the basal plane, and carboxyl groups at plane edges.13 Graphite oxide consists of loosely bound layers. Each layer has a two-dimensional arrangement of carbon atoms in the graphene pattern with a thickness of approximately 1.1 nm.14 The oxygen-containing functional groups can easily fall off from the surface of graphite oxide through calcination in inert gas atmosphere because of its special structure. Meanwhile, graphite oxide is exfoliated and turns into graphene oxide, reduced graphene oxide (rGO), or graphene based on different calcination temperatures. The present heterogeneous catalysts Co3O4/GO are fabricated through supporting cobalt oxide on the surface of graphene oxide. Calcination may significantly influence the catalytic performance of Co3O4/GO, but the correlated research is insufficient.
In this study, we fabricated Co3O4/GO nanocomposites as a heterogeneous catalyst for activation of PMS, which was used in the degradation of Orange II in water by AOPs-SRs. We calcinated Co3O4/GO nanocomposites at different temperatures of 300 °C, 500 °C, 700 °C, and 900 °C for 2 h in N2 atmosphere. As the calcination temperature was increased, the catalytic activity of the catalysts changed accordingly. The relationship between structure and catalytic activity was investigated via some physical characterization. We highlighted the influence of calcination temperature on the catalytic activity of Co3O4/GO catalysts, which has not been investigated before.
2. Experimental
2.1. Materials
Flake graphite (325 mesh, 99.99%) was supplied by Shanghai Yifan Graphite Co. Ltd, China. PMS, available as a triple salt of sulfate commercially known as Oxone (2KHSO5·KHSO4·K2SO4, 4.5% to 4.9% active oxygen), was obtained from Shanghai Ansin Chemical Co. Ltd and used as an oxidant. Orange II (98% purity) was supplied by Shanghai Yiji Dye Chemicals. Other reagents including Co(NO3)2·6H2O, H2SO4 (98%), NaNO3, KMnO4, and H2O2 (30%) of analytical grade were provided by Sinopharm Chemical Reagent Co. Ltd. (China).
2.2. Preparation of catalysts
GO is exfoliated graphite oxide. Graphite oxide was prepared from purified natural graphite with a mean particle size of 300 mesh according to the method reported by Hummers and Offeman.5,15 Graphite oxide (50 mg) was dispersed into 30 mL of hexyl alcohol through sonication for 3 h. The suspension was centrifuged (below 4000 rpm) to remove the sediment, and the supernatant liquid was stored for further use. Meanwhile, 0.25 mmol Co(NO3)2·6H2O was dissolved into another 10 mL of hexyl alcohol. The mixture was magnetically stirred for 2 h, and the resulting mixture was heated to 140 °C under reflux and vigorous magnetic stirring for 12 h. After the system was cooled to room temperature, the suspension was centrifuged, washed with absolute ethanol and water several times until all the remaining hexyl alcohol and other sundries were removed, and dried in a vacuum oven at 60 °C for 24 h. The product was labeled as Co3O4/GO. Bare GO and pure Co3O4 were synthesized with the same parameters for comparison. Finally, the dried Co3O4/GO was calcined in tube furnace at 300 °C, 500 °C, 700 °C, and 900 °C for 2 h in a flowing N2 atmosphere.
2.3. Evaluation of catalytic activity
The degradation experiments were conducted in a 250 mL vessel at room temperature. In a typical reaction of Orange II degradation with 0.6 mM, 0.05 g L−1 of catalyst and 6 mM of Oxone were added. Samples were obtained at regular intervals, joined immediately with equal methanol as quencher,2 and then filtered. Orange II concentration in the filtrate was measured in a UV-vis spectrophotometer (Shimadzu 2550) at 486 nm, and make full spectrum scan from 200–700 nm at the same time. Commercial Oxone can be used to generate PMS, and 614.7 g L−1 of Oxone is necessary to release 2 M of PMS. For the best results of degradation, the experiment was conducted at neutral (pH 7.0, adjusted with 0.5 M phosphate buffer) conditions.5 And most of the experiments were conducted in triplicate.
2.4. Characterization and analytical techniques
The structural features and the mineralogy of the catalysts were studied using X-ray diffraction (XRD) patterns obtained on a RIGAKU XRD instrument, using filtered CuKα radiation with accelerating voltage of 40 kV and current of 200 mA. The sample was scanned at 2 from 5° to 90°.
Thermogravimetric analysis (TGA) was conducted by a NETZSCH simultaneous thermal analyzer TG 209. About 10 mg samples were load in an alumina pan, and then heated from 25 to 900 °C at a ramping rate of 10 °C min−1 under N2 atmosphere. The vacant alumina pan was used as a reference throughout the conduct.
The Raman spectrum was acquired on a LabRAM HR Evolution (Horiba Scientific, France) with a Raman shift from 800 to 2000 cm−1 using an excitation wavelength of 532 nm diode laser excitation on a 300 lines per mm grating at room temperature.
The nanoscale structures were observed using high resolution transmission electron microscopy (FE-HRTEM, JEOL JEM-2100F) with an accelerating voltage of 200 kV. The sample was prepared by dispersing a small amount of dry powder in ethanol or water. Then, one droplet of the suspension was dropped on a formvar-carbon-coated, 300 mesh copper TEM grids (Ted Pella) covered with thin amorphous carbon film and allowed to evaporate in air at room temperature.
The atomic composition of the catalysts was detected by X-ray photoelectron spectroscopy (XPS). The XPS spectrum was recorded on a ESCALAB 250 photoelectron spectrometer (Thermo-VG Scientific, USA) with Al Kα (1486.6 eV) as the X-ray source. All XPS spectra were corrected using the C 1s line at 284.6 eV.
3. Results and discussion
3.1. Catalysts characterization
To understand the calcination temperature effect on the structural features and the mineralogy of the catalysts, Co3O4/GO calcined at 300 °C, 500 °C, 700 °C, and 900 °C were determined via XRD. Fig. 1 shows the wide angle XRD profiles of uncalcined and calcined samples of Co3O4/GO. As shown in Fig. 1, in the case of each sample, the patterns show reflection planes (111), (220), (311), (400), (511), and (440) at 18.96°, 31.20°, 36.74°, 44.72°, 59.20°, and 65.10°, respectively. The patterns indicate the presence of spinel Co3O4.16 These diffraction lines provide clear evidence of the existence of Co3O4 nanoparticles in the samples, which is in good agreement with the Joint Committee on Powder Diffraction Standards (JCPDS) card 43-1003.17 The different crystallinity degrees of Co3O4 were enhanced depending on the increasing calcination temperature. No diffraction peaks related to a Co2O3 or CoO phase were detected, which indicates that only Co3O4 was present in the uncalcined catalysts. Compared with uncalcined catalysts, the calcined catalysts had several characteristic diffraction peaks. In all the calcined catalysts, diffraction peaks related to CoO were observed. Five diffraction peaks at 36.48°, 42.38°, 61.48°, 73.66°, and 77.52° can be assigned to reflection planes (111), (200), (220), (311), and (222) of CoO crystal lattice, respectively, which is in good agreement with the JCPDS card 43-1004.18 As the calcination temperature was increased, the intensity of CoO diffraction peaks gradually increased, which indicates a high crystallinity. In addition, when the calcination temperature was increased to 700 °C, three new diffraction peaks were observed at 44.3°, 51.2°, and 75.8° related to metallic Co, which is in good agreement with the JCPDS card 15-0806.19 Meanwhile, as the calcination temperature was increased from 700 °C to 900 °C, the crystallinity of Co became more obvious. On the other hand, the typical diffraction peak of graphite oxide (001) is weak in the uncalcined sample, which indicates that the graphite oxide was mostly exfoliated to the GO sheet. On the other hand, the additional broad diffraction peak at 2 of 20° to 27° can be also indexed to the disorderedly stacked GO sheets.20 Calcination leads to the exfoliation of graphite oxide and the disappearance of the (001) diffraction peak. GO sheets can be reduced when calcined at high temperature. As the calcination temperature was increased, the samples displayed a broader graphitic (002) and weaker (100) peak.21 The presence of these peaks imply that the interplanar carbon bonds of the pristine graphite are broken, and graphene nanosheets are formed.22
 |
| | Fig. 1 XRD spectra of Co3O4/GO without calcination and calcined at 300, 500, 700 and 900 °C, respectively. | |
Fig. 2 shows the weight loss (TG) and the associated derivative thermogram (DTG) curves of Co3O4/GO, which had been dried at 60 °C for 24 h. This figure shows that the TG of the sample underwent continuous decline when the temperature was increased from ambient temperature to 900 °C. The most obvious stage starts from around 30 °C to 280 °C, where a mass loss of about 15% was observed. This step can be ascribed to dehydration of the compound and several reactions in the sample. Meanwhile, some oxygen-containing functional groups on the surface of GO sheet were removed, and GO was reduced to reduced GO or graphene. As the temperature was increased to 900 °C, no more obvious stage was observed except for a continuous decline. This phenomenon indicated that the component of Co3O4/GO reacted continually. The reaction may have occurred as eqn (1)–(7). When the temperature was increased to greater than 200 °C, the reduction reaction of Co3O4 occurred as follows:
| |
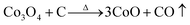 | (1) |
| |
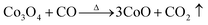 | (2) |
| |
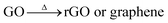 | (3) |
when the temperature rose to about 700 °C, reduction reaction occurred in-depth,
| |
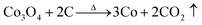 | (4) |
| |
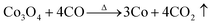 | (5) |
| |
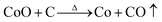 | (6) |
| |
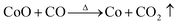 | (7) |
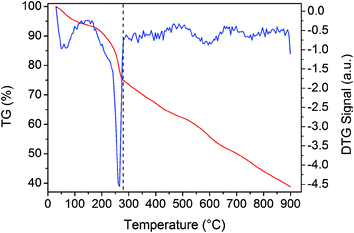 |
| | Fig. 2 TG and DTG curves of Co3O4/GO. | |
The result is consistent with the XRD analyses.
Raman spectroscopy is a powerful nondestructive tool used to distinguish ordered and disordered crystal structures of carbonaceous materials. The G band is common to all sp2 carbon forms and provides information on the in-plane E2g phonon of sp2 bonded carbon atoms, whereas the D band is a breathing mode of the k-point phonons of A1g symmetry.23 The D/G intensity ratio (ID/IG) is a measure of disorder degree and average size of the sp2 domains. Fig. 3 shows the Raman spectra of Co3O4/GO uncalcined, calcined at 500 °C and 900 °C. All of them exhibited a strong D band at ∼1346−1. However, uncalcined Co3O4/GO exhibited a G band at ∼1600 cm−1, while the corresponding G bands of Co3O4/GO calcined at 500 °C and 900 °C are ∼1575 cm−1. The red shifts of G band can be attributed to the high ability for recovery of the hexagonal network of carbon.24 The ID/IG of uncalcined Co3O4/GO was about 1.04, whereas the ID/IG of Co3O4/GO calcined at 500 °C and 900 °C were 0.92 and 1.33. Compared with uncalcined Co3O4/GO, the decreased ID/IG for Co3O4/GO calcined at 500 °C indicating the removal of oxygen-containing functionalities because of calcination.25 As the calcination temperature was increased, the ID/IG for Co3O4/GO calcined at 900 °C gradually increased because of the emission of CO2, CO and H2O (eqn (1)–(7)), which resulting a large number of carbon cavities and the increased disordered degree.26 Meanwhile, the introduction of CoO and Co nanoparticles (eqn (1)–(7)) make the RGO or graphene sheets monodisperse to keep large active specific surface area.27 This phenomenon also suggested an increase in the average size of the sp2 domains upon conversion from GO to RGO or graphene (eqn (3)), which in turn can be explained if newly created graphite domains are smaller in size than the ones present in GO.28 The results were consistent with the XRD and TG analyses.
 |
| | Fig. 3 Raman spectra of Co3O4/GO uncalcined, calcined at 500 °C and 900 °C. | |
A detailed microstructural study was performed by TEM to complete the characterization of the uncalcined samples and those calcined at high temperatures on the nanometer scale. The TEM images of uncalcined and calcined Co3O4/GO at 500 °C and 900 °C are shown in Fig. 4. Some Co3O4 nanoparticles were distributed on the surface of GO sheet in the uncalcined Co3O4/GO (Fig. 4a). The average particle size of Co3O4 is about 13.75 nm, which was calculated using the (311) diffraction peak of Co3O4 on the surface of GO by the Scherrer formula.29 After Co3O4/GO was calcined at 500 °C in N2 atmosphere, some nanoparticles were fairly homogeneously dispersed on the surface of the laminated structure (GO, rGO or graphene) (Fig. 4b). The nanoparticles could be Co3O4 and CoO based on the XRD analyses. When the calcination temperature was increased to 900 °C, the nanoparticles were also uniformly distributed and the mean grain size of the nanoparticles on the surface of the laminated structure was smaller than the size of the nanoparticles in Co3O4/GO calcined at 500 °C (Fig. 4c). This result is due to the generation of metallic Co caused by the reduction of Co3O4 or CoO. This result is consistent with the XRD and TG analyses. Fig. 4d shows the high-magnification of Co3O4/GO calcined at 900 °C. As shown in Fig. 4d, the particles on the surface of the laminated structure had a lattice spacing of 0.467 nm, which corresponds to the (311) planes of Co3O4. The lattice fringes at 0.213 nm of the particle with a softer contrast correspond to the (200) planes of CoO. Particularly, the particle with a lattice spacing of 0.203 nm corresponds to the (111) planes of Co, which indicates the generation of metallic Co. All these characterization results point to a progressive reduction of Co3O4 to CoO and Co at the reaction conditions.30,31
 |
| | Fig. 4 TEM images of Co3O4/GO uncalcined (a), calcined at 500 °C (b), 900 °C with low (c) and high-magnification (d). | |
The surface element compositions, metal oxidation states, and adsorbed species of solid materials were investigated via XPS. The XPS spectra of survey, C 1s, O 1s, and Co 2p in the uncalcined and calcined catalysts at 900 °C are shown in Fig. 5. The binding energies obtained in the XPS analyses were corrected for specimen charging by referencing the C 1s peak to 284.6 eV. The uncalcined and calcined catalysts at 900 °C have different spectra for both C 1s, O 1s, and Co 2p. The survey spectra are illustrated in Fig. 5a, which indicates that the XPS spectra of the uncalcined and calcined catalysts at 900 °C are similar. Moreover, both spectra have C 1s, O 1s, Co 2p, Co LMM, O KLL, Co 2s, Co 3s, Co 3p, and O 2s. The C 1s XPD spectra of the catalysts are shown in Fig. 5b. The C 1s XPD spectra of the uncalcined catalyst can be deconvoluted into a considerable degree of oxidation with four components that correspond to carbon atoms in different functional groups as follows: carbon in C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–H at 284.6 eV, carbon in C–O at 285.3 eV, C–O–C at 286.2 eV, and CO at 288.5 eV. On the other hand, the C 1s XPD spectra of the calcined catalyst at 900 °C only present two peaks at 284.6 and 286.2 eV, which correspond to the graphite structure and epoxy carbon. The disappearance of the C–O and CO peaks in the XPS spectrum indicates that some or all of the epoxide, hydroxyl, and carboxyl functional groups were removed because of calcination, which means GO was partially or fully reduced to rGO or graphene.32 The result is very consistent with those obtained from XRD, TG and TEM analyses.
C and C–H at 284.6 eV, carbon in C–O at 285.3 eV, C–O–C at 286.2 eV, and CO at 288.5 eV. On the other hand, the C 1s XPD spectra of the calcined catalyst at 900 °C only present two peaks at 284.6 and 286.2 eV, which correspond to the graphite structure and epoxy carbon. The disappearance of the C–O and CO peaks in the XPS spectrum indicates that some or all of the epoxide, hydroxyl, and carboxyl functional groups were removed because of calcination, which means GO was partially or fully reduced to rGO or graphene.32 The result is very consistent with those obtained from XRD, TG and TEM analyses.
 |
| | Fig. 5 XPS spectra of catalysts uncalcined and calcined at 900 °C: (a) survey scan, (b) C 1s region, (c) O 1s region, (d) Co 2p region. | |
As shown in Fig. 5c, the O 1s spectra of both uncalcined and calcined at 900 °C catalysts are clearly asymmetric, which indicates the existence of different oxygen species on the surface.33 In the O 1s pattern of uncalcined catalyst, three O 1s sub peaks are resolved with binding energies at 533.0, 532.0, and 531.3 eV, which are assigned to the oxygen in adsorbed water molecules, the hydroxyl oxygen (C–OH) in GO, and the cobalt hydroxide (Co–OH) on the surface, respectively.34 On the other hand, the O 1s pattern of the calcined catalysts at 900 °C is distinctively different. Aside from the main peak at 531.2 eV that corresponds to the Co–OH on the surface, a shoulder at a binding energy of 529.7 eV is attributed to the lattice oxygen species from cobalt oxide because of the hygroscopic moisture before testing. Compared with the uncalcined catalytic, the calcined catalysts at 900 °C had no XPS peak at 532.0 eV and but had a peak at 529.7 eV, which indicates that the C–OH disappeared and the lattice oxygen species from cobalt oxide appeared in the hybrid. Meanwhile, the subdued XPS peak at 533.0 eV indicates a decrease in the amount of adsorbed water molecules in the hybrid.
Fig. 5d shows the XPS spectra of Co 2p in the uncalcined and calcined catalysts at 900 °C. Each spectrum has two major peaks with binding energies at 781.1 or 779.8 eV and 797.3 or 795.4 eV, which correspond to Co 2p3/2 and Co 2p1/2, respectively. Both spectra have two corresponding shake-up satellite peaks located at approximately 6 eV above the main peaks, which is characteristic of the Co3+ and Co2+ phase.35,36 Compared with the catalyst calcined at 900 °C, the uncalcined catalyst had an XPS peak that is at a higher binding energy, which indicates a stronger interaction between the graphite support and the cobalt oxide species.37 Meanwhile, compared with those of Co 2p3/2 (778.10 eV) and Co 2p1/2 (793.30 eV) of metallic Co, a small chemical shift toward higher binding energies occurred for Co2p3/2 (779.8 eV) and Co2p1/2 (795.4 eV) (Fig. 5d), which indicates that the metallic Co on the surface of the graphene sheets was oxidized.38 Both XPS peaks at 780.7 eV for the uncalcined catalyst and 778.0 eV for are attributed to the surface Co3+ species and the peaks at 781.8 and 780.3 eV are attributed to the surface Co2+ species.39 Meanwhile, as shown in Fig. 5d, the XPS peak of Co3+ species in the catalyst calcined at 900 °C is weaker than that of uncalcined catalyst. On the other hand, the XPS peak of Co2+ species is stronger. This phenomenon indicates an increase in CoO and a decrease of Co3O4 in the calcined catalyst. This observation is in good agreement with the XRD, TG, Raman and TEM results.
3.2. Catalytic activities
The degradation curves of Orange II using Co3O4/GO of the uncalcined and calcined at 300 °C, 500 °C, 700 °C, and 900 °C as catalysts via AOPs-SRs are shown in Fig. 6. All experiments were done at neutral conditions (adjusted with 0.5 M phosphate buffer). The pH value during the whole reaction process was almost constant. As shown in Fig. 6, when the calcination temperature was increased, the catalytic activity of the catalyst obviously declined and then increased when the calcination temperature was above 700 °C. Moreover, the catalyst calcined at 900 °C had the best catalytic activity. This phenomenon indicates that calcination results in a significant change in the catalytic activity of the catalysts. The uncalcined Co3O4/GO had a high catalytic activity, which indicates that calcination at low temperatures (<500 °C) has an inhibitory effect on catalyst. This result indicated that the structure of Co3O4/GO was changed along with the change in calcination temperature, which leads to the change in catalytic activity. Although many experts demonstrated that the activity of the catalyst is greatly influenced by the increase in calcination temperature,7,40–42 no universally accepted standard is available because of the complexity of the catalyst compound.
 |
| | Fig. 6 Degradation curves of Orange II by using Co3O4/GO as a catalyst. | |
In order to better illustrate the degradation efficiency, the temporal evolution of UV-vis spectra of the Orange II solution were test. The initial UV-vis spectra of Orange II solution was shown in Fig. 7a, three characteristic peaks at 230, 255 and 310 nm were response to aromatic rings (benzene and naphthalene ring). The characteristic peak at 403 nm was ascribed to the NN group of azo form. The shoulder at 485 nm was due to the transition n–ð (the hydroxyl group toward the nitrogen bridge of hydrazone form).43 The temporal evolution of UV-vis spectra of the Orange II over uncalcined and calcined at different temperature were shown in Fig. 7b and c. As the experiment went on, the two characteristic peaks at 403 and 485 nm decreased simultaneously, suggesting that the two tautomeric forms react at the same time. Finally, the two characteristic absorption peaks were disappeared. Fig. 7d reported the UV-vis spectra of the Orange II over Co3O4/GO (uncalcined, calcined at 300 °C, 500 °C, 700 °C and 900 °C) at 2 min. It is more overtly demonstrated that the result is in good agreement with the degradation curves of Orange II (Fig. 6).
 |
| | Fig. 7 UV-vis spectra of Orange II over Co3O4/GO: (a) initial UV-vis spectra of the Orange II solution, (b) temporal evolution of UV-vis spectra of the Orange II over uncalcined (b1), calcined at 300 °C (b2), 500 °C (b3) and 700 °C (b4); (c) temporal evolution of UV-vis spectra of the Orange II over calcined at 900 °C; (d) the UV-vis spectra of the Orange II over different catalyst at 2 min. | |
3.3. Mechanism analysis
Many reports are available on the mechanism of the catalytic activity of heterogeneous catalysts in the degradation of organic pollutants. Many studies attribute the catalytic reaction to the formation of Co–OH on the surface of catalysts because the catalytic reaction starts with eqn (8)–(10).2,6,27,44–46 The formation of Co–OH complexes is a critical step for radical generation in the heterogeneous activation of PMS. In the presence of cobalt ions, PMS breaks up to generate active sulfate radicals via electron transfer mechanism from cobalt cations (eqn (8)–(10)). Meanwhile, the reverse electron transfer from Co3+ to Co2+ makes the reaction proceed cyclically until PMS is completely consumed at a sufficient reaction time.5,47,48 Experiments have shown that the uncalcined catalysts present high catalytic activity (Fig. 6).
In this work, the effect of calcination on catalytic activity was also studied. The effects of calcination on the catalyst structure are shown in Fig. 8. Calcination results in a change in the structure of the catalysts (eqn (1)–(7)). According to previous studies, calcination temperature has a key function in the catalytic activity of the catalyst. When the calcination temperature is increased, the catalytic activity of the catalysts first decreases and then increases. Compared with catalytic activity of the uncalcined catalyst, those of the catalysts calcined at 300 °C and 500 °C decrease mainly because of a decrease in the number of Co–OH on the surface of the catalyst compound, which was verified via XPS (Fig. 5). The formation of Co–OH is crucial for radical generation in the catalytic reaction (eqn (8)–(10)). Meanwhile, another reason is the decrease in Co3O4 and increase in CoO (eqn (1) and (2)). Cobalt redox cycling in the radical generation reaction takes place mainly in Co2O3 (contained in Co3O4), which is an unstable oxide.46 CoO is in dissolved form and did not appear to react heterogeneously in redox reactions. As such, the decrease in Co3O4 and increase in CoO in the catalysts result in the decrease in catalytic activity.
 |
| | Fig. 8 The effects of calcination on the catalyst structure. | |
When the calcination temperatures were further increased, the catalytic activity of the catalysts increased. Two aspects mainly contributed to the increase in catalytic activity. First, high-temperature calcination results in the exfoliation of GO and a decrease in oxygen-containing groups on the surface of GO, which lead to the formation of reduced GO (rGO) or even graphene. This phenomenon may result in the enhancement of catalytic activity following five factors:47 (i) rGO or graphene has an important function in the generation of SO4−˙ through the electron transfer between Co2+ and Co3+ because of its peculiar electronic structure and the high migration efficiency of electrons. (ii) rGO or graphene is beneficial to the homodisperse of nano-particles on its surface, which can enhance the catalytic activity of the hybrid. (iii) rGO or graphene with cobalt oxide on its surface can form strong interaction (Co–O–C), which favors the formation of Co–OH and promotes the activation of PMS to generate SO4−˙.6,49 (iv) rGO or graphene has extraordinary adsorption capacities because of its special two-dimensional structure, which can significantly increase the concentration of organic molecules near the catalytic surface and then enhance the catalytic degradation rates.50 (v) rGO or graphene also can activate PMS to produce SO4−˙ for the degradation of Orange II (eqn (11) and (12)). Therefore, the formation of reduced rGO or graphene because of calcination could enhance the catalytic activity of the hybrid.
Meanwhile, high temperature leads to the reduction of cobalt oxide to metallic cobalt (eqn (4)–(7)). In other words, when the catalyst is calcined at high temperatures, the product is Co/rGO or Co/graphene. At particular conditions, metallic cobalt (Co0) can release Co2+ (eqn (13) and (14)), which was employed to activate PMS to generate SO4−˙ for the degradation of Orange II in water (eqn (15)). Meanwhile, Co0 also can activate PMS to generate SO4−˙ (eqn (16)). PMS can also reduce Co3+ to Co2+ and generate SO5−˙ (eqn (17)). Thus, Co2+ exhibits redox cycling in the presence of PMS and produces SO4−˙.38 Furthermore, Co3+ is deposited on to the surface of Co0 to initiate the release of Co2+ (eqn (18)).38 In the presence of cobalt ions, PMS breaks up to generate active SO4−˙, which consequently aids in the oxidation of Orange II (eqn (19)).
The reaction mechanism is proposed as follows:
| | |
Co2+ + H2O ↔ CoOH+ + H+
| (8) |
| | |
CoOH+ + HSO5− → CoO+ + SO4−˙ + H2O
| (9) |
| | |
CoO+ + 2H+ ↔ Co3+ + H2O
| (10) |
| | |
rGO + HSO5− → rGO-H + SO5−˙
| (11) |
| | |
Graphene + HSO5− → graphene-H + SO5−˙
| (12) |
| | |
Co0 + O2 + 2H2O → Co2+ + 4OH−
| (14) |
| | |
Co2+ + HSO5− → Co3+ + SO4−˙ + OH−
| (15) |
| | |
Co0 + 2HSO5− → Co2+ + 2SO4−˙ + 2OH−
| (16) |
| | |
Co3+ + HSO5− → Co2+ + SO5−˙ + H+
| (17) |
| | |
SO4−˙ + Orange II → [… many steps …] → CO2 + H2O
| (19) |
4. Conclusions
Heterogeneous catalysts Co3O4/GO were prepared via solvothermal method. The catalysts were then calcined at 300 °C, 500 °C, 700 °C, and 900 °C and characterized via XRD, TG, Raman, XPS, and TEM. Both uncalcined and calcined catalysts showed high catalytic activity at high temperatures in degrading Orange II from water using AOPs-SRs. Calcination had a significant function in catalytic activity. The effects of calcination temperature on the morphology, atomic composition, and crystallite structure of the catalysts were investigated. The relationship between calcination temperature and catalytic activity was discussed. In accordance with the increase in calcination temperature, the number of Co–OH decreased. This decline, which resulted in weakened catalytic reaction, was crucial for radical generation in the catalytic reaction. At the same time, the amount of CoO in dissolved form increased and did not appear to react heterogeneously in redox reactions. When the calcination temperature was further increased, cobalt oxide was reduced to metallic cobalt, and GO was partially or fully exfoliated and reduced to rGO or graphene. The metallic cobalt can release Co2+ and activate PMS directly, and rGO or graphene has a high electron migration efficiency and can also activate PMS directly, all of which resulted in the enhancement of catalytic activity. In summary, the catalytic activity of Co3O4/GO can be considerably influenced by calcination temperature. More study on the effects of calcination temperature should be conducted for the preparation of highly efficient catalysts.
Acknowledgements
The study was supported by Shanghai Municipal Natural Science Foundation (no. 15ZR1417800), “Dawn” Program of Shanghai Education Commission (no. 11SG52), Shanghai Key Project for Fundamental Research (no. 13JC1402800), Science and Technology Commission of Shanghai Municipality (no. 14DZ2261000), and Scientific Research Foundation of Shanghai University of Electric Power (no. K2014-015).
References
- H. V. Lutze, R. Bakkour, N. Kerlin, C. Sonntag and T. C. Schmidt, Water Res., 2014, 53, 370 CrossRef CAS PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Appl. Catal., B, 2004, 54, 155 CrossRef CAS PubMed.
- J. E. Mondloch, E. Bayram and R. G. Finke, J. Mol. Catal. A: Chem., 2012, 355, 1 CrossRef CAS PubMed.
- P. H. Shi, R. J. Su, F. Z. Wan, M. C. Zhu, D. X. Li and S. H. Xu, Appl. Catal., B, 2012, 123–124, 265 CrossRef CAS PubMed.
- P. H. Shi, X. F. Dai, H. A. Zheng, D. X. Li, W. F. Yao and C. Y. Hu, Chem. Eng. J., 2014, 240, 264 CrossRef CAS PubMed.
- M. T. Makhlouf, B. M. Abu-Zied and T. H. Mansoure, Appl. Surf. Sci., 2013, 274, 45 CrossRef CAS PubMed.
- Y. B. Yu, J. J. Zhao, X. Han, Y. Zhang, X. B. Qin and B. Y. Wang, Chin. J. Catal., 2013, 34, 283 CrossRef CAS.
- P. Broqvist, I. Panas and H. Persson, J. Catal., 2002, 210, 198 CrossRef CAS.
- Q. S. Guo, X. Y. Guo and Q. H. Tian, Adv. Powder Technol., 2010, 21, 529 CrossRef CAS PubMed.
- S. C. Petitto, E. M. Marsh, G. A. Carson and M. A. Langell, J. Mol. Catal. A: Chem., 2008, 281, 49 CrossRef CAS PubMed.
- S. Royer and D. Duprez, ChemCatChem, 2011, 3, 24 CrossRef CAS PubMed.
- D. Chen, H. B. Feng and J. H. Li, Chem. Rev., 2012, 112, 6027 CrossRef CAS PubMed.
- K. Zhang, V. Dwivedi, C. Y. Chi and J. S. Wu, J. Hazard. Mater., 2010, 182, 162 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- T. Yu, Y. Zhu, X. Xu, Z. Shen, P. Chen, C. Lim, J. T. Thong and C. H. Sow, Adv. Mater., 2005, 17, 1595 CrossRef CAS PubMed.
- T. Battumur, S. B. Ambade, R. B. Ambade, P. Pokharel, D. S. Lee, S. H. Han, W. J. Lee and S. H. Lee, Curr. Appl. Phys., 2013, 13, 196 CrossRef PubMed.
- J. S. Do and C. H. Weng, J. Power Sources, 2005, 146, 482 CrossRef CAS PubMed.
- J. L. Qiao, L. Xu, L. Ding, L. Zhang, R. Baker, X. F. Dai and J. J. Zhang, Appl. Catal., B, 2012, 125, 197 CrossRef CAS PubMed.
- Z. S. Wu, W. C. Ren, L. Wen, L. B. Gao, J. P. Zhao, Z. P. Chen, G. M. Zhou, F. Li and H. M. Cheng, ACS Nano, 2010, 4, 3187 CrossRef CAS PubMed.
- Y. S. Fu, H. Q. Chen, X. Q. Sun and X. Wang, Appl. Catal., B, 2012, 111–112, 280 CrossRef CAS PubMed.
- Y. Q. He, Y. Liu, T. Wu, J. K. Ma, X. R. Wang, Q. J. Gong, W. N. Kong, F. B. Xing, Y. Liu and J. P. Gao, J. Hazard. Mater., 2013, 260, 796 CrossRef CAS PubMed.
- F. Tuinstra and J. L. Koening, J. Chem. Phys., 1970, 53, 1126 CrossRef CAS PubMed.
- D. Kang, J. Y. Kwon, H. Cho, J. Sim, H. S. Hwang, C. S. Kim, Y. J. Kim, R. S. Ruoff and H. S. Shin, ACS Nano, 2012, 6, 7763 CrossRef CAS PubMed.
- J. Yan, T. Wei, W. Qiao, B. Shao, Q. Zhao, L. Zhang and Z. Fan, Electrochim. Acta, 2010, 55, 6973 CrossRef CAS PubMed.
- B. Zhao, G. H. Zhang, J. S. Song, Y. Jiang, H. Zhuang, P. Liu and T. Fang, Electrochim. Acta, 2011, 56, 7340 CrossRef CAS PubMed.
- G. M. Zhou, D. W. Wang, F. Li, L. L. Zhang, N. Li, Z. S. Wu, L. Wen, G. Q. Lu and H. M. Cheng, Chem. Mater., 2010, 22, 5306 CrossRef CAS.
- S. C. Ray, A. Saha, S. K. Basiruddin, S. S. Roy and N. R. Jana, Diamond Relat. Mater., 2011, 20, 449 CrossRef CAS PubMed.
- H. Borchert, E. V. Shevchenko, A. Robert, I. Mekis, A. Kornowski, G. Grübel and H. Weller, Langmuir, 2005, 21, 1931 CrossRef CAS PubMed.
- Y. Wang, H. J. Zhang, L. Lu, L. P. Stubbs, C. C. Wong and J. Y. Lin, ACS Nano, 2010, 4, 4753 CrossRef CAS PubMed.
- J. Llorca, P. R. Piscina, J. A. Dalmon and N. Homs, Chem. Mater., 2004, 16, 3573 CrossRef CAS.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 8, 1 Search PubMed.
- Q. J. Yang, H. Choi and D. D. Dionysiou, Appl. Catal., B, 2007, 74, 170 CrossRef CAS PubMed.
- F. Ahmed and D. F. Rodrigues, J. Hazard. Mater., 2013, 256–257, 33 CrossRef CAS PubMed.
- J. Yan, T. Wei, W. M. Qiao, B. Shao, Q. K. Zhao, L. J. Zhang and Z. J. Fan, Electrochim. Acta, 2010, 55, 6973 CrossRef CAS PubMed.
- S. L. Xiong, C. Z. Yuan, M. F. Zhang, B. J. Xi and Y. T. Qian, Chem.–Eur. J., 2009, 15, 5320 CrossRef CAS PubMed.
- J. F. Chen, Y. R. Zhang, L. Tan and Y. Zhang, Ind. Eng. Chem. Res., 2011, 50, 4212 CrossRef CAS.
- Y. J. Yao, C. Xu, J. C. Qin, F. Y. Wei, M. N. Rao and S. B. Wang, Ind. Eng. Chem. Res., 2013, 52, 17341 CrossRef CAS.
- M. M. Natile and A. Glisenti, Chem. Mater., 2003, 15, 2502 CrossRef CAS.
- N. M. Deraz, Mater. Lett., 2002, 57, 914 CrossRef CAS.
- J. B. Li, Z. Q. Jiang, K. Qian and W. X. Huang, Chin. J. Chem. Phys., 2012, 25, 103 CrossRef CAS.
- V. Balouria, S. Samanta, A. Singh, A. K. Debnath, A. Mahajan, R. K. Bedi, D. K. Aswal and S. K. Gupta, Sens. Actuators, B, 2013, 176, 38 CrossRef CAS PubMed.
- D. Chen, X. L. Ma, J. Z. Zhou, X. Chen and G. R. Qian, J. Hazard. Mater., 2014, 279, 476 CrossRef CAS PubMed.
- X. Y. Chen, J. W. Chen, X. L. Qiao, D. G. Wang and X. Y. Cai, Appl. Catal., B, 2008, 80, 116 CrossRef CAS PubMed.
- W. Zhang, H. L. Tay, S. S. Lim, Y. H. Wang, Z. Y. Zhong and R. Xu, Appl. Catal., B, 2010, 95, 93 CrossRef CAS PubMed.
- G. P. Anipsitakis, E. Stathatos and D. D. Dionysiou, J. Phys. Chem. B, 2005, 109, 13052 CrossRef CAS PubMed.
- Y. J. Yao, Z. H. Yang, H. Q. Sun and S. B. Wang, Ind. Eng. Chem. Res., 2012, 51, 14958 CrossRef CAS.
- Q. Y. Yan, X. Y. Li, Q. D. Zhao and G. H. Chen, J. Hazard. Mater., 2012, 209–210, 385–391 CrossRef CAS PubMed.
- G. P. Anipsitakis, D. D. Dionysiou and M. A. Gonzalez, Environ. Sci. Technol., 2006, 40, 1000 CrossRef CAS.
- S. D. Perera, R. G. Mariano, K. Vu, N. Nour, O. Seitz, Y. Chabal and K. J. Balkus, ACS Catal., 2012, 2, 949 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra02680f |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 














