DOI:
10.1039/C5RA02365C
(Paper)
RSC Adv., 2015,
5, 25967-25978
Design, synthesis and biological evaluation of novel 1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolins as potential EZH2 inhibitors
Received
6th February 2015
, Accepted 4th March 2015
First published on 4th March 2015
Abstract
The histone lysine methyltransferase EZH2 has been implicated as a key component in cancer aggressiveness, metastasis and poor prognosis. This study discovered a new class of hexahydroisoquinolin derivatives as EZH2 inhibitors. A structure–activity relationship study showed that the steric hindrance was important to the activity for EZH2. A preliminary optimization study led to the discovery of several potent compounds with low nanomolar to sub-nanomolar potency for EZH2. Biological evaluation indicated that SKLB1049 was a highly potent with improved solubility compared to EPZ6438, SAM-competitive, and cell-active EZH2 inhibitor that decreased global H3K27me3 in SU-DHL-6 and Pfeiffer lymphoma cells in a concentration- and time-dependent manner. Further study indicated that SKLB1049 caused cell arrest in G0/G1 phase. These compounds would be useful as chemical tools to further explore the biology of EZH2 and provided us with a start point to develop new EZH2 inhibitors.
Introduction
Cancer is a major public health problem in the world.1 Targeting epigenetic regulators has provided us with a new strategy for cancer therapy.2 Enhancer of Zeste Homolog 2 (EZH2) is a member of the histone lysine methyltransferase family and the catalytic component of the polycomb repressive complex 2 (PRC2) as well.3 PRC2 catalyzes the methylation of histone 3 lysine 27 (H3K27), leading to trimethylation of H3K27 (H3K27me3), which is an epigenetic mark of transcriptionally repressive and silences target genes.4 The over expression of EZH2 has been observed in numerous blood and solid tumors, such as Hodgkin's disease,5 non Hodgkin's lymphoma,6 prostate,7 breast,8 renal,9 melanoma10 and tongue11 cancers, and is widely implicated in cancer progression, metastasis and poor prognosis.8,12 Recently, somatic mutations of tyrosine Y641, Alanine A677 and A687 of EZH2 were reported to be associated with germinal center B-cell like diffuse large B-cell lymphoma (GCB-DLBCL) and Follicular Lymphoma (FL).13–15 Thus, inhibition of EZH2 is considered as an attractive therapeutic target for the treatment of cancer.
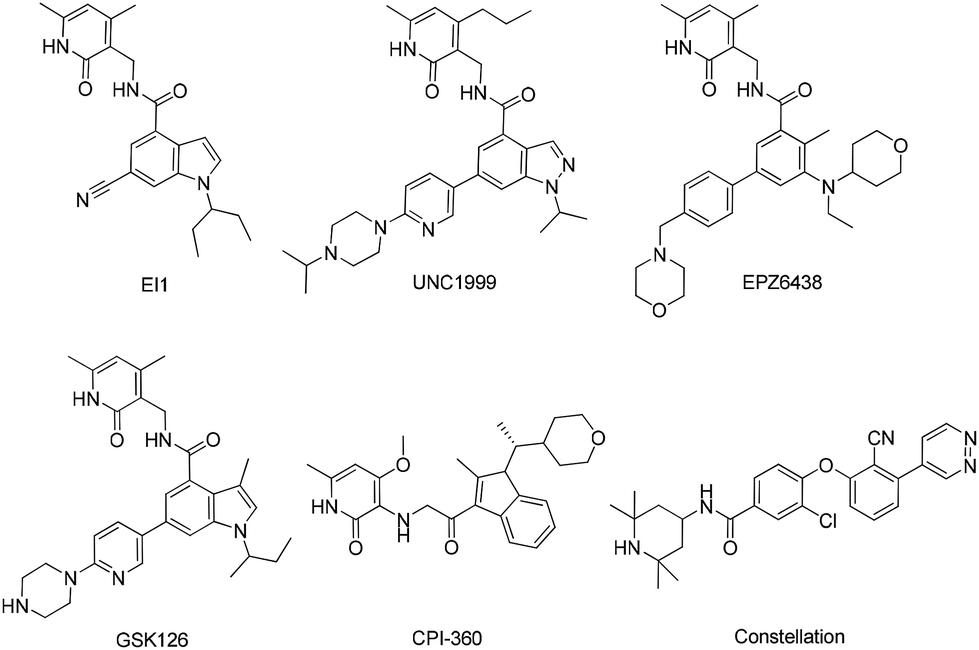
Over the recent years, multiple groups, including GlaxoSmithKline,16 Epizyme,17–19 Novartis,20 UNC Eshelman21 and Constellation,22,23 have reported various structures of small molecule inhibitors of EZH2 (Fig. 1). The most potent compound EPZ6438 was reported to inhibit EZH2 with an IC50 of 2.5 nM, and exhibited significant tumor growth inhibition in several human DLBCL17 and malignant rhabdoid18 xenograft models. In 2013, a phase I/II study of EPZ6438 for the treatment of advanced solid tumors and B cell lymphomas was initiated. However, the effective dose of EPZ6438, 600 mg kg−1 once a day, was very high in the WSU-DLCL2 xenograft model, which could attribute to its inferior physicochemical properties, such as solubility. Thus highly potent EZH2 inhibitor with improved physicochemical properties is emergently needed.
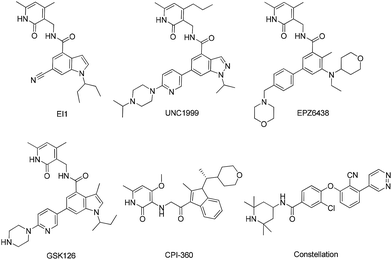 |
| | Fig. 1 Disclosed EZH2 inhibitors. | |
Our research group designed and synthesized a series of novel EZH2 inhibitors bearing hexahydroisoquinolins, and the potency of these compounds for EZH2 was characterized. Biological evaluation indicated that SKLB1049 was highly potent for EZH2 methyltransferase with improved solubility than EPZ6438. In cell-based assays, SKLB1049 remarkably decreased global H3K27me3 levels, and selectively killed two DLBCL cell lines harboring the EZH2 mutant, SU-DHL-6 and Pfeiffer cells, but no apparent impact on normal cell lines. Further study indicated that SKLB1049 caused cell cycle arrest in G0/G1 phase. This study may contribute to the further design of novel scaffolds against EZH2.
Results and discussion
Chemistry
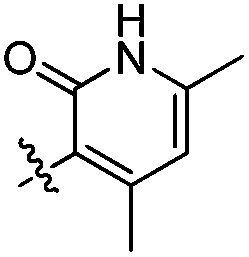
The compounds described in this study were synthesized according to Scheme 1. Briefly, treatment of ketone 1 with ethyl acetate under sodium hydride24 or acetyl chloride under LDA25 afforded 1,3-diones 2. Then the key building blocks pyridine-2-(1H)-ones 3 were prepared by treating 1,3-diones with cyanoacetamide in the presence of triethylenediamine (DABCO) in refluxing ethanol.26 Subsequently, reduction of 3 by sodium borohydride and iodine afforded amines 4,27 which were subjected to coupling reaction with various acids 8 afforded 9.28 Next, the title compounds 10 were gained by Suzuki coupling reaction with various boronic acids or boronic esters. Compound 7 used for preparation of acid 8 was synthesized under a two-step reductive amination reaction, starting from methyl 3-amino-5-bromo-2-methylbenzoate 5. The structures of all the tested compounds were fully characterized by 1H NMR, 13C NMR and ESI-MS analysis.
 |
| | Scheme 1 Reagents and conditions: (a) NaH, EA, PhMe, 0 °C-RT or LDA, acetyl chloride, THF, −78 °C; (b) cyanothioacetamide, DABCO, ethanol, reflux; (c) NaBH4, I2, THF, 0 °C-reflux; (d) sodium triacetoxyborohyride, acetic acid, trichloromethane, 0 °C-RT; (e) sodium triacetoxyborohyride, acetic acid, 1,2-dichloroethane, 0° C-RT; (f) 3 M NaOH, ethanol, RT, 3 h, then adjust to pH 4–5 with 1 N HCl; (g) EDC, HOAT, NMM, DMSO, RT, 12–48 h; (h) PdCl2(dppf)·CH2Cl2, Na2CO3, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dioxane–H2O, 110 °C. :1 dioxane–H2O, 110 °C. | |
SAR analysis
Modifications of 4- and 5-positions of pyridone moiety. Tables 1 and 2 provided the biological data of these compounds. All synthesize derivatives were evaluated for its EZH2 inhibitory activity in AlphaLISA29 assay and the antiproliferation activities of SU-DHL-6 and Pfeiffer cells in MTT assay. To investigate the structure–activity relationships of C4 and C5 position of pyridone moiety, we first synthesized compounds 10a–c wherein pyridone C4–C5 position together formed an alkyl ring. Evaluation of the ring size of the fused system demonstrated that the seven- and eight-membered rings (10b and 10c) were equipotent and inferior to the six-membered ring (10a, EZH2WT IC50 = 14 nM; EZH2Y641 IC50 = 27.2 nM; EZH2A677G IC50 = 7.0 nM), which was chosen for further SAR study. Simple methyl substitution at the 7-position of the terminal fused ring (10d) improved the potency on all three EZH2 enzymes: EZH2WT, EZH2Y641F and EZH2A677G, while the ethyl (10e), tert-butyl (10f) and dimethyl analogs (10g) were less active. This revealed that a relative small substituent in this position was more potent, and the volume of the substitutents was a crucial factor for the activity. Introduction of a methyl to C6 position of 1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin resulted in a less potent compound (10h). Comparison of 10h and 10d suggested that methyl substituted at the 7-position of the hexahydroisoquinoline was preferred relative to the 6-position. Interestingly, introduction of a phenyl ring fused to cyclohexane moiety led to an inactive compound 10i, which suggested that a bulky substituent in C5 and C6 of hexahydroisoquinoline might change its conformation and was harmful to the anti-EZH2 activity. In this series of pyridone analogs (10a–10i), 10d displayed the most active for EZH2 inhibition. However, it did not show remarkably enhanced antiproliferation activities for SU-DHL-6 and Pfeiffer cells compared with 10a in this 4 days assay.
Table 1 In vitro potencies of newly synthesized inhibitors 10a–10i
Table 2 In vitro potencies of newly synthesized inhibitors 10j–10u
Substitutions of R2 position of central phenyl. Subsequent SAR study of R2 position of central phenyl was further explored with a series of aryl and heteroaryl (Table 2). For this purpose, the fused cyclohexane group was held constant. The different R2 substituents had considerable influence on the activity for EZH2. Among aryl substitutuents, compound 10q was the most potent compound with an IC50 of 7.3 nM for EZH2WT, which was 2-fold improvement compared with 10a. On the other hand, replacement of the 4-(morpholinomethyl) phenyl moiety (10a) with 1-methyl-1H-pyrazole-4-yl (10r), 2-fluoropyridin-5-yl (10s) or 2-chloropyridin-5-yl (10t) retained excellent potency for EZH2, and introduction of 2-piperazinyl pyridin-5-yl (10u, EZH2WT IC50 = 4.0 nM) in this position led to a 4-fold improvement of potency. In this series (10a, 10j–10u), most compounds did not show obvious effect on tumor cells viability. However, 10q and 10u displayed slightly improved antiproliferative activities for SU-DHL-6 and Pfeiffer cells compared with 10a.
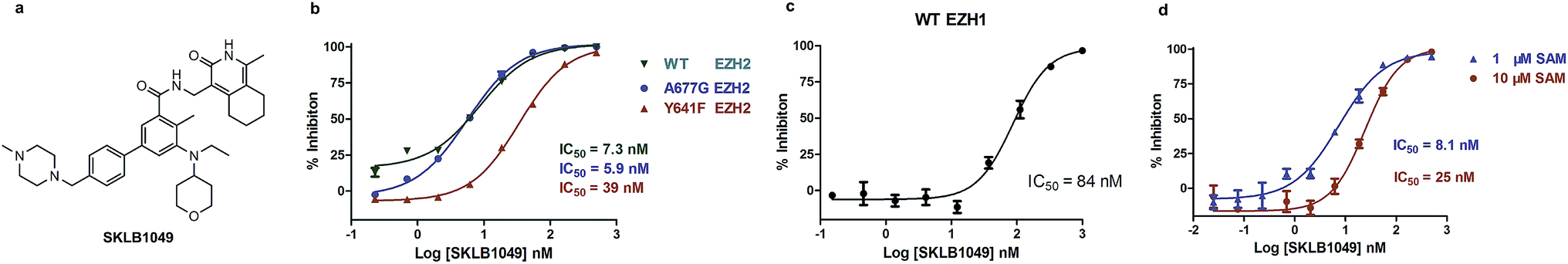
SKLB1049 was a SAM-competitive, potent, and selective inhibitor of EZH2. On the basis of the high enzyme and cells inhibitory activities and improved solubility, SKLB1049 was selected for subsequent studies for mechanism of action (MOA), selectivity and cellular assays. To elucidate the MOA of EZH2 inhibition by SKLB1049, the IC50 values were determined at two different SAM concentrations of 1 μM and 10 μM. As shown in Fig. 2d, the results demonstrated that the increase of SAM concentration could cause an obvious shift of IC50 value of SKLB1049 against EZH2WT, which indicated that SKLB1049 was a SAM-competitive EZH2 inhibitor. The MOA for SKLB1049 was consistent with that of structurally similar molecule EPZ6438.
 |
| | Fig. 2 SKLB1049 was a potent, SAM-competitive and selective inhibitor of EZH2. (a) Chemical structures of SKLB1049. (b) Determination of the inhibitory potential of SKLB1049 on WT EZH2 (green), Y641F mutant EZH2 (red), and A677G EZH2 (blue). (c) Determination of the inhibitory potential of SKLB1049 on WT EZH1. (d) Determination of the mechanism of action of SKLB1049. Data were represented as mean values ± SEM (n = 2). | |
As shown in Fig. 2b, SKLB1049 was highly potent for both Y641F and A677G mutants, with IC50 values of 39 nM and 5.9 nM, respectively. The potency of SKLB1049 for WT enzymes was similar with A677G mutant and superior to Y641F mutant. To determine the selectivity profile of SKLB1049, the potency for EZH1 was also evaluated (Fig. 2c). SKLB1049 showed a considerable potency for EZH1, with an IC50 value of 84 nM, approximately 12-fold less potent than EZH2.
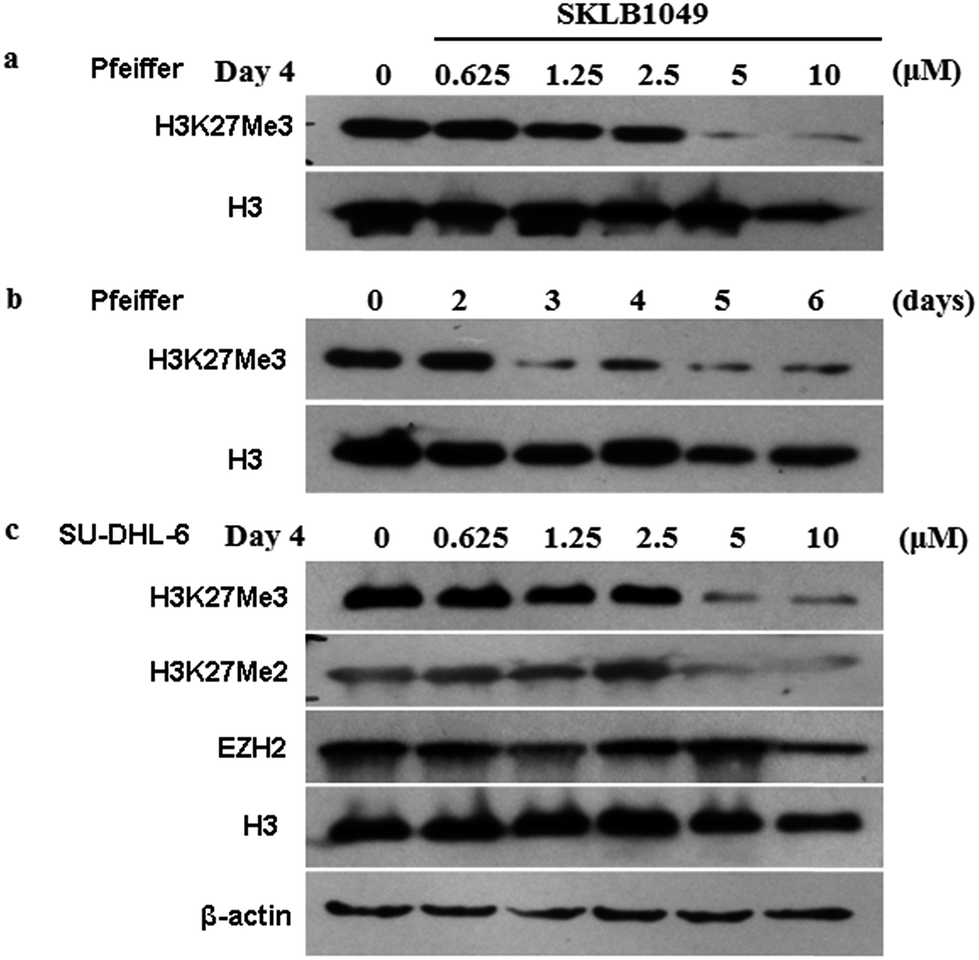
SKLB1049 potently reduced H3K27me3 in cells. The ability of SKLB1049 to reduce H3K27me3 in intact cells was evaluated in SU-DHL-6 and Pfeiffer cells using Western blotting analysis (Fig. 3). Treatment with SKLB1049 at 5 μM for 4 days and 6 days potently abolished H3K27me3 mark in EZH2 A677G mutant Pfeiffer cells in a concentration- and time-dependent manner. Similarly, SKLB1049 remarkably reduced H3K27me3 and H3K27me2 mark in a concentration-dependent manner in SU-DHL-6 cells, with no affection on EZH2 levels.
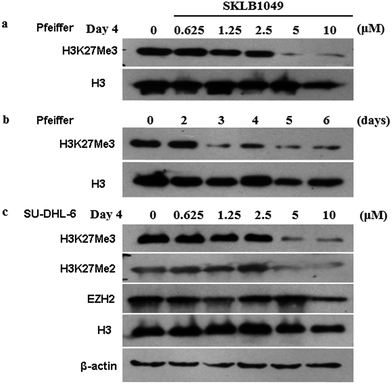 |
| | Fig. 3 Compound SKLB1049 reduced H3K27me3 in Pfeiffer and SU-DHL-6 cells in a dose- and time-dependent manner. (a) Pfeiffer cells was treated with 0.625 μM, 1.25 μM, 2.5 μM, 5 μM or 10 μM of SKLB1049 for 4 days and whole cell extracts were analyzed by western blotting for levels of H3K27me3. H3 and β-actin levels served as loading controls. (b) Similarly effect of H3K27me3 levels with 5 μM SKLB1049 in Pfeiffer cells over 6 days were determined. (c) SU-DHL-6 cells was treated of SKLB1049 for 4 days and effect of H3K27me3, H3K27me2 and EZH2 levels were determined. H3 and β-actin levels served as loading controls. | |
SKLB1049 inhibited proliferation of cancer cell line. To further investigate the effect of EZH2 inhibition on cell proliferation, the inhibitory activities of 10a, SKLB1049 and 10u against SU-DHL-6 and Pfeiffer cell lines for 6 days were evaluated (Table 4). As shown in Fig. 4, cell lines were effectively killed by EZH2 inhibitors in a time-dependent manner. SKLB1049 and 10u demonstrated slightly greater potency than 10a in both cell lines. This was consistent with the better activity of SKLB1049 and 10u on EZH2 activity. Pfeiffer was more sensitive to EZH2 inhibition compared with SU-DHL-6, with IC50 values of 2.76 μM, 1.19 μM and 1.65 μM for 10a, SKLB1049 and 10u, respectively. The difference in sensitivity between Pfeiffer and SU-DHL-6 was not due to different levels of target engagement because the similar significant suppression of global H3K27me3 in both cell lines was observed.
Table 4 The MTT data (6 days)a
| Compound |
IC50 (μM) |
| SU-DHL-6 |
Pfeiffer |
HEK-293 |
LO2 |
VERO |
| IC50 value was average of three determinations and deviation from the average was <5% of the average value. |
| 10a |
10.02 |
2.76 |
>40 |
>40 |
>40 |
| SKLB1049 |
6.17 |
1.19 |
>40 |
>40 |
>40 |
| 10u |
2.65 |
1.65 |
>40 |
>40 |
>40 |
| EPZ6438 |
6.12 |
1.47 |
>40 |
>40 |
10.81 |
 |
| | Fig. 4 Viability across SU-DHL-6 and Pfeiffer cell lines treated with 10a, SKLB1049 and 10u for 4 and 6 days was assessed. Data were represented as mean values ± SEM (n = 3). | |
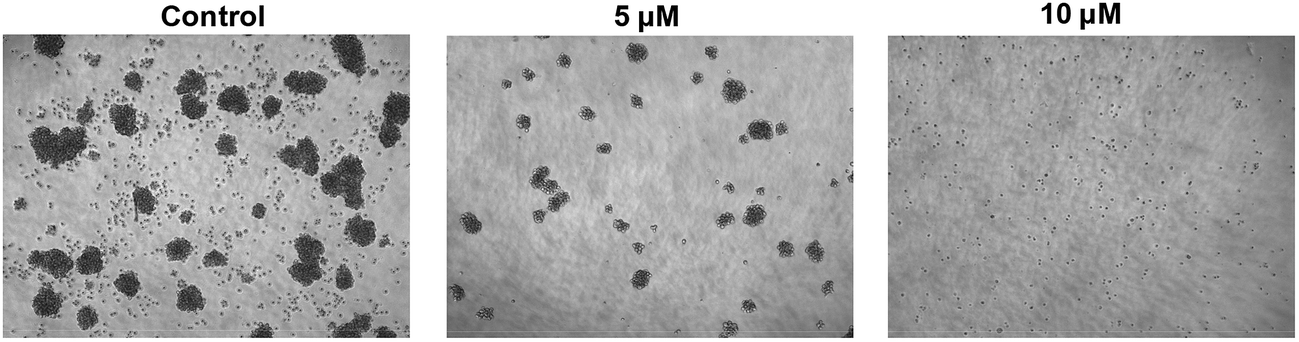
Then, bright-field microscopy images of SU-DHL-6 cancer cells after incubation with SKLB1049 for 6 days at varying concentrations were performed to assess the activity visually. As shown in Fig. 5, when treated with SKLB1049 for 6 days, the proliferation of SU-DHL-6 cancer cell was remarkably inhibited, and these phenomena were more significant as the concentration increasing.
 |
| | Fig. 5 Bright-field microscopy images of SU-DHL-6 cancer cells, after incubation with SKLB1049 for 6 days at varying concentrations: 5 μM, 10 μM. | |
To further evaluate the safety of our EZH2 inhibitors, the antiproliferation activities of SKLB1049 for 6 days against three normal cell lines, HEK 293, Vero and LO2 were measured by MTT assay (Table 4). All these compounds showed no obvious impact on these normal cells, indicating its good in vitro safety profile.
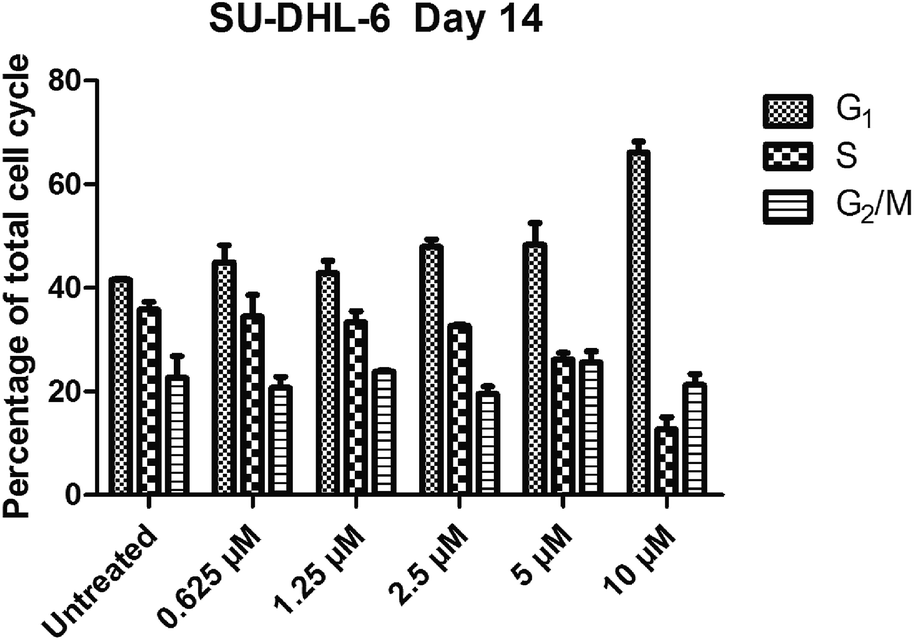
SKLB1049 could remarkably arrested G0/G1 phase. The effect on cell cycle progression of SKLB1049 was also assessed (Fig. 6). Treatment with SKLB1049 in SU-DHL-6 cells at a concentration of 0.625 μM showed no change in cell cycle compared with the DMSO control, while a concentration of 10 μM treatment resulted in an increase of the percentage of cells in G0/G1 phase from 44.89% to 66.09%, and a concomitant decrease in S phase from 34.43% to 12.68%. However there was no considerable change in the percentage of cells in the G2/M phase through 14 days treatment. These results suggested that SKLB1049 could remarkably arrested cell cycle in G0/G1 phase in a concentration-dependent manner.
 |
| | Fig. 6 Compound SKLB1049 induced G0/G1 arrest in EZH2 mutant SU-DHL-6 cells. Cell cycle analysis (by flow cytometry) was assessed in SU-DHL-6 cells during incubation with either vehicle or 0.625 μM, 1.25 μM, 2.5 μM, 5 μM, 10 μM SKLB1049 for 14 days. Data were represented as mean values ± SEM (n = 2). | |
Conclusions
We designed and synthesized a series of novel compounds bearing hexahydroisoquinolin and their biological activities of targeting EZH2 were characterized. Structure–activity relationship study showed the steric hindrace was important to the activity for EZH2. Preliminary optimization led to the discovery of several potent compounds with good solubility, in which SKLB1049 was selected for subsequent studies for mechanism of action (MOA), selectivity and cellular assays. Biological evaluation indicated that SKLB1049 was a highly potent candidate for EZH2 wild-type, Y641 mutant and A677 mutant enzymes with improved solubility than EPZ6438, through a SAM-competitive manner. In cell-based assays, SKLB1049 remarkably reduced global H3K27me3 and H3K27me2 levels in a concentration- and time-dependent manner, with no affection on EZH2 levels. SKLB1049 could selectively killed two DLBCL cell lines harboring the EZH2 mutants, SU-DHL-6 and Pfeiffer cells in a concentration- and time-dependent manner, but with no apparent impact on normal cell lines. Further study indicated that SKLB1049 caused cell arrest in G0/G1 phase. Together, our study may contribute to the further design of novel scaffolds against EZH2. Further studies of the biological mechanisms and anti-tumor research in vivo are in progress.
Experimental
Chemistry
Unless otherwise noted, all materials were obtained from commercial suppliers and used without purification. The 1H and 13C NMR spectra were recorded on a Bruker AVANCE III 400 spectrometer using DMSO-d6 or CDCl3 as the solvent. Chemical shifts (δ) were reported in ppm relative to Me4Si (internal standard), and coupling constants (J) were reported in Hz. Mass Spectra (MS) were performed on a Waters Q-TOF Premier mass spectrometer. Thin layer chromatography (TLC) used Qingdao Haiyang Silica gel F-254 plates, and column chromatography were performed using Qingdao Haiyang Silica gel 60 (300–400 mesh). HPLC analysis was performed on an UltiMate 3000 HPLC system (Dionex, USA). All tested compounds were purified until the purity was ≧95%, detected by HPLC under UV 254 nm wavelength, NMR and ESI-MS.
General procedure for the preparation of 1,3-diketones 2a–2h
Method A for 2a–2b, 2e–2h. A mixture of cyclic ketone (14 mmol, 1.0 equiv) and toluene (0.5 mL) was added dropwise to a suspension of 60% sodium hydride (28 mmol, 2.0 equiv) in ethyl acetate (28 mmol, 2.0 equiv). After hydrogen evolution ceased, the reaction mixture was heated at 40 °C for 3 h. Additional stirring at room temperature was continued and excess sodium hydride was quenched with methanol. The reaction mixture was poured into water, neutralized with concentrated HCl, and extracted with CH2Cl2. The combined extracts were washed with brine, dried, and concentrated. Purification by silica flash chromatography (hexanes/EtOAc) giving 2a, 2b, 2e, 2e. Yeild: 30–60%.
Method B for 2c–2d. A solution of cyclic ketone (1.0 equiv) in dry THF (0.67 M) was added to a stirred solution of freshly prepared LDA (lithium diisopropylamide) (1.0 equiv) in THF (1.0 M) at −78 °C. The resulting solution was stirred for 2 h. A solution of acetyl chloride (1.0 equiv) in THF (1.2 M) was added to the above solution. After stirring at −78 °C was continued for 1 h, the cooling bath was removed and stirring was continued for 3 h. The reaction mixture was poured into a saturated aqueous NH4Cl (25 mL) and then extracted with CH2Cl2 (3×). The combined extracts were washed with brine, dried, and concentrated. Purification by silica flash chromatography (hexanes/EtOAc) giving 2c, 2d. Yeild: 50–60%.
General procedure for the synthesis of 3
1,3-Diketones (14 mmol, 1.0 equiv), cyanoacetamide (14 mmol, 1.0 equiv) and DABCO (14 mmol, 1.0 equiv) was added to a solution of EtOH (25 mL), then the resulting mixture was stirred at 90 °C for 10 h. The reaction mixture was cooled to rt and the solid precipitate was filtered, washed with EtOH and dried to afford the desired compound. Yeild: 20–60%.
General procedure for the synthesis of 4
To an ice-bath cooled THF (100 mL) solution of 3 (10 mmol, 1.0 equiv) were added NaBH4 (11 mmol, 1.1 equiv) and I2 (10 mmol, 1.0 equiv) and the mixture stirred for 30 min. The reaction mixture was then heated at reflux for 10 h and then allowed to cool to room temperature. After cooling to 0 °C, the reaction mixture was acidified by slow addition of 3 N HCl (l mL). The reaction mixture was concentrated in vacuo and the crude product 4 used directly in the next step without further purification.
General procedure for the synthesis of 6
The starting material methyl 3-amino-5-bromo-2-methylbenzoate (5) was synthesized based on a literature method. To a stirred solution of 5 (10 mmol, 1.0 equiv) and cyclic ketone (15 mmol, 1.5 equiv) in dichloroethane (50 mL) was added acetic acid (60 mmol, 6.0 equiv) and the reaction mixture stirred at room temperature for 15 minutes, then the reaction mixture was cooled to 0 °C and sodium triacetoxyborohydride (30 mmol, 3.0 equiv) was added. The reaction mixture was stirred overnight at room temperature. Upon completion of the reaction as determined by TLC, aqueous sodium bicarbonate solution was added to the reaction mixture until a pH of 7–8 was obtained. The organic phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography eluting with ethyl acetate: hexane to afford the desired compound 6 as a solid. Yeild: 70%.
General procedure for the synthesis of 7
To a stirred solution of 6 (10 mmol, 1.0 equiv) and acetaldehyde (15 mmol, 1.5 equiv) in dichloroethane (50 mL) was added acetic acid (60 mmol, 6.0 equiv) and the reaction mixture stirred at room temperature for 15 minutes, then the reaction mixture was cooled to 0 °C and sodium triacetoxyborohydride (30 mmol, 3.0 equiv) was added. The reaction mixture was stirred overnight at room temperature. Upon completion of the reaction as determined by TLC, aqueous sodium bicarbonate solution was added to the reaction mixture until a pH of 7–8 was obtained. The organic phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography eluting with ethyl acetate: hexane to afford the desired compound 7 as a liquid. Yeild: 60%.
General procedure for the synthesis of 8
Ester 7 (10 mmol, 1.0 equiv) in ethanol (25 mL) was added aqueous NaOH (15 mmol, 1.5 equiv in 5 mL water) and the resulting mixture was stirred at 60 °C for 1 h. Upon completion of the reaction as determined by TLC, the solvent was removed under reduced pressure and the residue obtained was acidified with 1 N HCl until a pH 4 was obtained. The precipitate was filtered off, washed with water and dried to afford the desired acid 8. Yeild: 80%.
General procedure for the synthesis of 9
The above acid 8 (1.0 mmol, 1.0 equiv) and amino 4 (2 mmol, 2.0 equiv) were dissolved in DMSO (10 mL), then HOAT (1.5 mmol, 1.5 equiv), EDCI (1.5 mmol, 1.5 equiv) and N-methylmorpholine (3 mmol, 3 equiv) were added to it. The reaction mixture was stirred at room temperature for 20 h. Upon completion of the reaction as determined by TLC, the reaction mixture was poured onto ice-cold water (100 mL), stirred for 30 minutes and the precipitated solid was collected by filtration, washed with water (50 mL) and air dried. The crude compound was purified by column chromatography eluting to afford the desired compound 9 as a solid. Yeild: 65–78%.
General procedure for the synthesis of 10
To a stirred solution of 9 (1.0 mmol, 1.0 equiv) in dioxane–water mixture (20 mL/2 mL) was added boronic ester derivatives (1.08 mmol, 1.08 equiv) followed by addition of Na2CO3 (3.6 mmol, 3.6 equiv). The solution was purged with argon for 15 minutes and then PdCl2(dppf)·CH2Cl2 (0.1 mmol, 0.1 equiv) was added and the solution was again purged with argon for a further 10 min. The reaction mixture was heated at 100 °C for 4 h. After completion (monitored by TLC), the reaction mixture was diluted with water and extracted with 10% MeOH/DCM. The combined organic layers were dried over anhydrous sodium sulphate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography eluting with methanol: DCM to the title compound as a solid.
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4′-(morpholinomethyl)biphenyl-3-carboxamide (10a). Yield: 85%; 1H NMR (400 MHz, DMSO) δ 11.50 (s, 1H), 8.17 (s, 1H), 7.58 (d, J = 6.8 Hz, 2H), 7.38 (m, 3H), 7.22 (s, 1H), 4.32 (s, 2H), 3.83 (d, J = 9.6 Hz, 2H), 3.58 (s, 4H), 3.49 (s, 2H), 3.25 (t, J = 10.9 Hz, 2H), 3.08 (m, 2H), 3.02 (m, 1H), 2.75 (s, 2H), 2.37 (s, 6H), 2.25 (s, 3H), 2.10 (s, 3H), 1.65 (s, 6H), 1.53 (m, 2H), 0.84 (s, 3H). 13C NMR (100 MHz, DMSO) δ 169.03, 161.53, 150.04, 148.84, 140.62, 139.60, 138.56, 136.95, 132.56, 129.48, 126.37, 122.78, 120.76, 111.76, 66.29, 66.16, 62.04, 57.81, 53.14, 41.05, 34.57, 30.23, 26.60, 24.09, 22.15, 21.89, 15.93, 14.55, 12.67. ESI-MS m/z 613.2 (M + H)+.
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1-methyl-3-oxo-3,5,6,7,8,9-hexahydro-2H-cyclohepta[c]pyridin-4-yl)methyl)-4′-(morpholinomethyl)biphenyl-3-carboxamide (10b). Yield: 74%; 1H NMR (400 MHz, DMSO) δ 11.40 (s, 1H), 8.19 (s, 1H), 7.58 (d, J = 7.7 Hz, 2H), 7.39 (d, J = 4.0 Hz, 2H), 7.36 (s, 1H), 7.22 (s, 1H), 4.38 (d, J = 4.0 Hz, 2H), 3.83 (d, J = 10.6 Hz, 2H), 3.58 (s, 4H), 3.48 (s, 2H), 3.25 (t, J = 11.2 Hz, 2H), 3.16 (m, 2H), 3.02 (m, 1H), 2.65 (d, J = 9.4 Hz, 2H), 2.36 (s, 4H), 2.26 (s, 3H), 2.19 (s, 3H), 1.74 (s, 2H), 1.67 (m, 2H), 1.53 (m, 4H), 1.45 (s, 2H), 1.24 (s, 2H), 0.83 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.16, 163.55, 150.05, 149.57, 147.42, 139.30, 137.89, 133.62, 131.73, 131.34, 131.24, 130.16, 128.37, 128.25, 126.84, 123.98, 121.93, 121.15, 120.73, 67.33, 66.23, 62.44, 58.41, 53.06, 41.54, 37.07, 33.29, 31.89, 30.47, 29.70, 29.37, 27.45, 26.49, 25.63, 22.70, 20.79, 16.94, 14.78, 14.15, 12.84. ESI-MS m/z 627.2 (M + H)+.
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8,9,10-octahydrocycloocta[c]pyridin-4-yl)methyl)-4′-(morpholinomethyl)biphenyl-3-carboxamide (10c). Yield: 84%; 1H NMR (400 MHz, DMSO) δ 11.35 (s, 1H), 8.18 (t, J = 4.8 Hz, 1H), 7.57 (d, J = 8.0 Hz, 2H), 7.39 (d, J = 4.4 Hz, 2H), 7.36 (s, 1H), 7.21 (s, 1H), 4.37 (d, J = 4.6 Hz, 2H), 3.83 (d, J = 10.4 Hz, 2H), 3.57 (s, 4H), 3.48 (s, 2H), 3.25 (t, J = 11.3 Hz, 1H), 3.08 (m, 2H), 3.02 (m, 1H), 2.60 (m, 2H), 2.36 (s, 4H), 2.24 (s, 3H), 2.21 (s, 3H), 1.66 (m, 2H), 1.58 (s, 2H), 1.54 (d, J = 8.0 Hz, 2H), 1.39 (s, 2H), 1.27 (s, 2H), 0.83 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.16, 164.15, 149.94, 149.52, 144.99, 144.42, 139.41, 137.81, 133.72, 130.18, 126.94, 123.96, 122.71, 120.71, 117.96, 67.34, 66.19, 62.48, 58.40, 53.09, 41.51, 37.07, 31.93, 30.80, 30.47, 29.80, 29.71, 29.57, 26.18, 25.65, 25.52, 16.18, 14.75, 14.15, 12.83. ESI-MS m/z 641.2 (M + H)+.
N-((1,7-Dimethyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)biphenyl-3-carboxamide (10d). Yield: 79%; 1H NMR (400 MHz, DMSO) δ 11.49 (s, 1H), 8.16 (t, J = 4.4 Hz, 1H), 7.57 (d, J = 8.0 Hz, 2H), 7.45–7.26 (m, 1H), 7.21 (s, 1H), 4.31 (t, J = 4.0 Hz, 2H), 3.83 (d, J = 10.2 Hz, 2H), 3.64–3.53 (m, 4H), 3.48 (s, 2H), 3.25 (t, J = 11.2 Hz, 2H), 3.14–3.04 (m, 2H), 3.04–2.87 (m, 1H), 2.61 (m, 2H), 2.36 (s, 4H), 2.25 (s, 3H), 2.10 (s, 3H), 1.90 (m, 1H), 1.80 (m, 1H), 1.66 (m, 4H), 1.52 (m, 2H), 1.19 (m, 1H), 1.01 (d, J = 6.4 Hz, 3H), 0.83 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.17, 163.60, 150.66, 149.63, 140.58, 139.36, 137.71, 133.75, 130.28, 126.81, 123.92, 121.10, 120.76, 114.88, 67.33, 66.01, 58.39, 52.88, 41.50, 36.12, 33.48, 30.47, 30.39, 28.52, 27.37, 21.91, 16.58, 14.78, 12.84. ESI-MS m/z 627.2 (M + H)+.
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-ethyl-1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4-methyl-4′-(morpholinomethyl)biphenyl-3-carboxamide (10e). Yield: 74%. 1H NMR (400 MHz, DMSO) δ 11.49 (s, 1H), 8.16 (s, 1H), 7.57 (s, 1H), 7.38 (s, 2H), 7.21 (s, 1H), 4.31 (s, 2H), 3.83 (s, 2H), 3.57 (s, 4H), 3.48 (s, 4H), 3.25 (s, 2H), 3.08 (s, 2H), 2.93 (m, 1H), 2.56 (s, 4H), 2.36 (s, 3H), 2.25 (s, 3H), 2.11 (s, 2H), 1.88 (m, 2H), 1.65 (m, 2H), 1.53 (m, 2H), 1.34 (m, 2H), 1.14 (m, 1H), 0.93 (s, 3H), 0.83 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.17, 163.60, 150.97, 149.64, 140.66, 140.56, 139.39, 133.79, 130.30, 126.80, 123.91, 121.05, 120.75, 114.85, 67.33, 58.39, 52.80, 41.48, 36.13, 35.16, 31.33, 30.48, 29.71, 29.38, 29.00, 28.00, 27.32, 22.71, 16.59, 14.78, 14.15, 12.84, 11.44. ESI-MS m/z 641.2 (M + H)+.
N-((7-tert-Butyl-1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)biphenyl-3-carboxamide (10f). Yield: 67%. 1H NMR (400 MHz, DMSO) δ 11.49 (s, 1H), 8.16 (t, J = 4.4 Hz, 1H), 7.57 (d, J = 8.0 Hz, 2H), 7.45–7.26 (m, 1H), 7.21 (s, 1H), 4.31 (t, J = 4.0 Hz, 2H), 3.83 (d, J = 10.2 Hz, 2H), 3.64–3.53 (m, 4H), 3.48 (s, 2H), 3.25 (t, J = 11.2 Hz, 2H), 3.14–3.04 (m, 2H), 3.04–2.87 (m, 1H), 2.61 (m, 2H), 2.36 (s, 4H), 2.25 (s, 3H), 2.10 (s, 3H), 1.90 (m, 1H), 1.80 (m, 1H), 1.66 (m, 4H), 1.52 (m, 2H), 1.19 (m, 1H), 1.01 (s, 9H), 0.83 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, DMSO) δ 170.04, 159.80, 155.87, 142.77, 141.48, 140.30, 139.71, 138.87, 136.80, 129.17, 128.90, 128.52, 128.20, 127.99, 116.49, 114.00, 67.12, 67.08, 63.42, 54.66, 51.93, 46.90, 46.37, 36.13, 32.67, 29.95, 29.31, 28.02, 26.85, 24.74, 16.01, 15.61, 14.39, 12.58. ESI-MS m/z 669.2 (M + H)+.
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-N-((1,7,7-trimethyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)biphenyl-3-carboxamide (10g). Yield: 69%. 1H NMR (400 MHz, DMSO) δ 11.51 (s, 1H), 8.21 (s, 1H), 7.57 (s, 2H), 7.45–7.26 (m, 1H), 7.21 (s, 1H), 4.31 (t, J = 4.0 Hz, 2H), 3.83 (d, J = 10.2 Hz, 2H), 3.64–3.53 (m, 4H), 3.48 (s, 2H), 3.25 (t, J = 11.2 Hz, 2H), 3.14–3.04 (m, 2H), 3.04–2.87 (m, 1H), 2.61 (m, 2H), 2.36 (s, 4H), 2.25 (s, 3H), 2.10 (s, 3H), 1.90 (m, 1H), 1.80 (m, 1H), 1.66 (m, 4H), 1.52 (m, 2H), 1.19 (m, 1H), 1.1 (s, 6H), 0.83 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.18, 150.25, 149.62, 141.12, 140.84, 139.39, 133.80, 130.37, 126.85, 123.89, 120.75, 114.48, 67.33, 65.87, 58.37, 52.81, 41.47, 38.56, 36.25, 34.73, 30.46, 28.75, 28.01, 24.61, 16.65, 14.76, 12.84. ESI-MS m/z 641.2 (M + H)+.
N-((1,6-Dimethyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)biphenyl-3-carboxamide (10h). Yield: 80%. 1H NMR (400 MHz, DMSO) δ 11.49 (s, 1H), 8.15 (t, J = 4.8 Hz, 1H), 7.54 (m, 2H), 7.48–7.30 (m, 3H), 7.22 (s, 1H), 4.33 (d, J = 4.8 Hz, 2H), 3.83 (d, J = 10.0 Hz, 2H), 3.70–3.53 (m, 4H), 3.48 (s, 2H), 3.25 (t, J = 11.2 Hz, 2H), 3.09 (m, 2H), 3.04–2.90 (m, 2H), 2.36 (s, 6H), 2.25 (s, 3H), 2.10 (s, 3H), 1.81 (m, 1H), 1.66 (m, 3H), 1.52 (m, 2H), 1.21 (m, 2H), 1.01 (d, J = 6.4 Hz, 3H), 0.83 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.17, 163.53, 151.06, 149.56, 140.62, 139.90, 139.40, 137.90, 133.59, 130.05, 126.82, 123.92, 120.93, 120.78, 114.46, 77.39, 77.07, 76.75, 67.33, 66.32, 62.53, 58.41, 53.14, 41.51, 36.07, 35.78, 30.47, 29.71, 28.51, 24.77, 21.91, 16.62, 14.78, 12.85. ESI-MS m/z 627.2 (M + H)+.
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-N-((2-oxo-2,3,5,6-tetrahydrobenzo[f]isoquinolin-1-yl)methyl)biphenyl-3-carboxamide (10i). Yield: 74%. 1H NMR (400 MHz, DMSO) δ 11.65 (s, 1H), 8.33 (s, 1H), 7.92 (s, 1H), 7.58 (d, J = 7.6 Hz, 3H), 7.46 (t, J = 8.0 Hz, 3H), 7.30 (s, 3H), 7.25 (s, 1H), 4.47 (d, J = 4.0 Hz, 2H), 3.83 (d, J = 10.0 Hz, 2H), 3.57 (s, 4H), 3.48 (s, 2H), 3.25 (t, J = 11.3 Hz, 2H), 3.09 (d, J = 6.7 Hz, 2H), 3.02 (m, 1H), 2.80 (m, 2H), 2.64 (m, 2H), 2.36 (s, 4H), 2.28 (s, 6H), 1.66 (m, 2H), 1.53 (m, 2H), 1.09 (t, J = 7.0 Hz, 3H), 0.83 (t, J = 6.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.32, 164.21, 149.58, 149.47, 139.64, 139.26, 138.20, 137.74, 135.98, 133.10, 129.81, 129.50, 128.26, 128.01, 126.86, 124.58, 123.97, 123.19, 120.79, 116.36, 67.29, 66.70, 62.88, 58.38, 53.41, 41.45, 36.79, 30.44, 28.17, 22.81, 16.40, 14.79, 12.86. ESI-MS m/z 661.2 (M + H)+.
5-Bromo-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)benzamide (10j). Yield: 74%. 1H NMR (400 MHz, DMSO) δ 11.38 (s, 1H), 8.24 (s, 1H), 7.31 (s, 1H), 7.09 (s, 1H), 4.33 (d, J = 4.6 Hz, 2H), 3.83 (d, J = 10.0 Hz, 2H), 3.24 (t, J = 10.8 Hz, 2H), 3.09–2.97 (m, 2H), 2.97–2.83 (m, 1H), 2.72 (s, 2H), 2.38 (s, 2H), 2.15 (s, 3H), 2.09 (s, 3H), 1.59 (m, 6H), 1.51 (m, 2H), 0.79 (t, J = 6.8 Hz, 2H). 13C NMR (100 MHz, DMSO) δ 167.74, 161.55, 150.38, 141.24, 140.33, 132.96, 127.06, 124.75, 120.87, 117.38, 111.76, 66.09, 58.15, 40.75, 34.55, 29.83, 26.61, 24.20, 22.17, 16.00, 14.82, 12.42. ESI-MS m/z 516.1 (M + H)+.
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)biphenyl-3-carboxamide (10k). Yield: 77%. 1H NMR (400 MHz, CDCl3) δ 13.30 (s, 1H), 7.46 (d, J = 6.6 Hz, 2H), 7.36–7.21 (m, 5H), 4.57 (s, 2H), 3.94 (d, J = 10.2 Hz, 2H), 3.31 (t, J = 9.6 Hz, 2H), 3.10 (d, J = 6.4 Hz, 2H), 3.02 (m, 1H), 2.95 (s, 2H), 2.30 (s, 2H), 1.97 (s, 3H), 1.71 (m, 7H), 1.26 (s, 4H), 0.89 (t, J = 6.4 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 169.72, 163.52, 151.73, 140.90, 140.21, 139.50, 138.74, 128.80, 127.51, 126.88, 123.88, 120.97, 72.79, 67.23, 42.50, 31.94, 30.19, 29.71, 29.37, 27.43, 24.85, 22.71, 22.19, 16.49, 15.07, 14.15, 12.60. ESI-MS m/z 514.2 (M + H)+.
4′-Chloro-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)biphenyl-3-carboxamide (10l). Yield: 80%. 1H NMR (400 MHz, DMSO) δ 11.59 (s, 1H), 8.19 (s, 1H), 7.66 (s, 2H), 7.49 (d, J = 5.3 Hz, 2H), 7.42 (s, 1H), 7.25 (s, 1H), 4.33 (s, 2H), 3.82 (s, 2H), 3.25 (s, 2H), 3.05 (m, 3H), 2.75 (s, 2H), 2.37 (s, 2H), 2.27 (s, 3H), 2.10 (s, 3H), 1.64 (s, 6H), 1.53 (m, 2H), 0.83 (s, 3H).13C NMR (100 MHz, CDCl3) δ 169.79, 163.47, 151.54, 140.86, 139.64, 138.67, 137.35, 133.81, 133.52, 128.90, 128.09, 123.66, 121.38, 120.95, 115.38, 72.83, 67.24, 59.30, 42.19, 35.95, 31.93, 30.23, 29.70, 29.37, 27.43, 24.87, 22.70, 22.17, 16.51, 15.05, 14.15, 12.71. ESI-MS m/z 548.2 (M + H)+.
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4′-(trifluoromethyl)biphenyl-3-carboxamide (10m). Yield: 64%. 1H NMR (400 MHz, DMSO) δ 11.48 (s, 2H), 8.19 (s, 1H), 7.86 (m, 2H), 7.80 (m, 2H), 7.48 (s, 1H), 7.30 (s, 1H), 4.32 (s, 2H), 3.84 (d, J = 7.5 Hz, 2H), 3.26 (m, 2H), 3.11 (m, 2H), 3.03 (m, 1H), 2.75 (s, 2H), 2.38 (s, 2H), 2.27 (s, 3H), 2.10 (s, 3H), 1.65 (s, 6H), 1.53 (m, 2H), 0.84 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.57, 165.86, 165.24, 163.10, 151.87, 143.76, 140.96, 139.83, 137.26, 129.70, 129.46, 128.31, 127.40, 127.21, 125.73, 125.50, 123.93, 122.80, 120.86, 115.87, 67.18, 35.91, 31.94, 30.12, 29.71, 29.37, 27.45, 24.89, 22.71, 22.13, 22.09, 16.59, 15.17, 14.14, 12.58. ESI-MS m/z 582.2 (M + H)+.
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methoxy-4-methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)biphenyl-3-carboxamide (10n). Yield: 77%. 1H NMR (400 MHz, DMSO) δ 11.55 (s, 1H), 8.15 (t, J = 4.8 Hz, 1H), 7.55 (d, J = 8.8 Hz, 2H), 7.35 (s, 1H), 7.18 (s, 1H), 7.00 (d, J = 8.8 Hz, 2H), 4.32 (d, J = 4.4 Hz, 2H), 3.82 (d, J = 10.8 Hz, 2H), 3.78 (s, 3H), 3.24 (t, J = 11.2 Hz, 2H), 3.07 (m, 2H), 3.00 (m, 1H), 2.74 (s, 2H), 2.37 (s, 2H), 2.23 (s, 3H), 2.10 (s, 3H), 1.64 (s, 6H), 1.53 (m, 2H), 0.83 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, DMSO) δ 169.11, 161.52, 158.78, 150.38, 148.77, 139.54, 136.82, 132.20, 127.64, 122.36, 120.29, 114.29, 66.30, 57.80, 55.15, 35.10, 30.26, 26.60, 24.08, 22.15, 21.89, 15.93, 14.49, 12.66. ESI-MS m/z 555.2 (M + H)+.
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4′-morpholinobiphenyl-3-carboxamide (10o). Yield: 87%. 1H NMR (400 MHz, DMSO) δ 11.49 (s, 1H), 8.12 (t, J = 4.6 Hz, 1H), 7.49 (d, J = 8.0 Hz, 2H), 7.34 (s, 1H), 7.16 (s, 1H), 7.01 (d, J = 8.4 Hz, 2H), 4.31 (d, J = 4.4 Hz, 2H), 3.83 (d, J = 10.8 Hz, 2H), 3.79–3.68 (m, 4H), 3.25 (t, J = 11.2 Hz, 2H), 3.13 (m, 4H), 3.08 (m, 2H), 3.05 (s, 1H), 2.74 (s, 2H), 2.38 (s, 2H), 2.22 (s, 3H), 2.10 (s, 3H), 1.64 (m, 6H), 1.52 (m, 2H), 0.83 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.07, 163.26, 151.46, 150.59, 140.83, 138.17, 127.53, 120.83, 115.70, 115.21, 77.39, 77.07, 76.75, 67.17, 66.76, 49.10, 36.24, 30.11, 29.71, 27.41, 24.89, 22.25, 22.19, 16.57, 15.04, 12.65. ESI-MS m/z 599.0 (M + H)+.
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-3′-morpholinobiphenyl-3-carboxamide (10p). Yield: 70%; 1H NMR (400 MHz, DMSO) δ 11.47 (s, 1H), 8.13 (t, 1H), 7.36 (s, 1H), 7.30 (t, J = 8.0 Hz, 1H), 7.20 (s, 1H), 7.09 (s, 1H), 7.02 (d, J = 8.0 Hz, 1H), 6.94 (d, J = 8.0 Hz, 1H), 4.32 (d, J = 4.0 Hz, 2H), 3.83 (d, J = 12.0 Hz, 2H), 3.76 (s, 4H), 3.25 (t, J = 12.0 Hz, 2H), 3.17 (m, 4H), 3.12–3.05 (m, 1H), 3.02 (m, 1H), 2.75 (s, 2H), 2.38 (s, 2H), 2.24 (s, 3H), 2.10 (s, 3H), 1.64 (m, 6H), 1.59–1.43 (m, 2H), 0.83 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, DMSO) δ 169.10, 161.52, 151.60, 150.09, 148.71, 140.79, 139.53, 137.84, 132.50, 129.45, 123.00, 121.03, 120.70, 117.74, 114.25, 113.23, 111.51, 66.28, 66.11, 57.83, 48.45, 41.14, 34.56, 30.27, 26.63, 24.08, 22.16, 21.90, 15.92, 14.54, 12.70. ESI-MS m/z 599.0 (M + H)+.
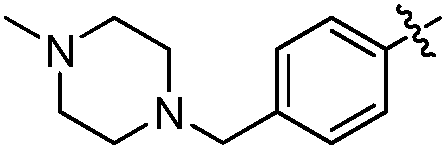
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4′-((4-methylpiperazin-1-yl)methyl)biphenyl-3-carboxamide (10q). Yield: 72%; 1H NMR (400 MHz, DMSO) δ 11.49 (s, 1H), 8.15 (s, 1H), 7.56 (d, J = 8.0 Hz, 2H), 7.48–7.28 (m, 3H), 7.22 (s, 1H), 4.32 (s, 2H), 3.83 (d, J = 8.4 Hz, 2H), 3.47 (s, 2H), 3.27 (m, 6H), 3.09 (m, 3H), 2.75 (s, 2H), 2.37 (s, 6H), 2.25 (s, 3H), 2.17 (s, 3H), 2.10 (s, 3H), 1.65 (m, 6H), 1.53 (m, 2H), 0.83 (s, 3H). 13C NMR (100 MHz, DMSO) δ 169.04, 161.52, 150.04, 148.84, 139.59, 138.49, 136.96, 132.53, 129.38, 126.35, 122.77, 120.75, 111.51, 66.29, 61.59, 57.81, 54.53, 52.28, 45.45, 34.57, 30.24, 26.60, 24.09, 22.15, 21.89, 15.93, 14.54, 12.67. ESI-MS m/z 626.2 (M + H)+.
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(1-methyl-1H-pyrazol-4-yl)-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)benzamide (10r). Yield: 60%; 1H NMR (400 MHz, CDCl3) δ 12.53 (s, 1H), 7.65 (s, 1H), 7.51 (s, 1H), 7.36 (s, 1H), 7.20 (s, 1H), 7.17 (s, 1H), 4.58 (d, J = 5.2 Hz, 2H), 3.94 (d, J = 10.4 Hz, 2H), 3.86 (s, 3H), 3.31 (t, J = 10.0 Hz, 2H), 3.07 (d, J = 6.8 Hz, 2H), 2.95 (m, 3H), 2.38 (s, 2H), 2.32 (s, 3H), 2.10 (s, 3H), 1.73 (m, 4H), 0.87 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, DMSO) δ 169.19, 161.55, 150.03, 148.73, 140.38, 139.69, 135.89, 130.76, 129.64, 127.78, 121.54, 121.24, 120.71, 119.02, 111.54, 66.31, 57.80, 41.08, 38.54, 34.55, 30.28, 26.60, 24.09, 22.14, 21.89, 15.93, 14.46, 12.71. ESI-MS m/z 518.1 (M + H)+.
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(6-fluoropyridin-3-yl)-2-methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)benzamide (10s). Yield: 85%; 1H NMR (400 MHz, DMSO) δ 11.51 (s, 1H), 8.53 (s, 1H), 8.27 (t, J = 7.2 Hz, 1H), 8.18 (s, 1H), 7.48 (s, 1H), 7.28 (s, 1H), 7.26 (s, 1H), 4.32 (d, J = 4.0 Hz, 2H), 3.83 (d, J = 10.5 Hz, 2H), 3.25 (t, J = 11.2 Hz, 2H), 3.10 (d, J = 6.4 Hz, 2H), 3.03 (m, 1H), 2.75 (s, 2H), 2.37 (s, 2H), 2.21 (s, 3H), 2.10 (s, 3H), 1.64 (s, 6H), 1.52 (m, 2H), 0.82 (t, J = 6.4 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 169.60, 164.35, 163.12, 161.88, 158.63, 151.99, 148.49, 145.63, 145.45, 140.94, 139.95, 139.62, 134.18, 133.96, 123.72, 120.96, 115.86, 109.66, 109.39, 67.20, 59.49, 42.25, 35.85, 30.20, 27.46, 24.92, 22.41, 16.50, 15.12, 12.91. ESI-MS m/z 533.0 (M + H)+.
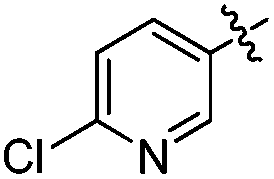
5-(6-Chloropyridin-3-yl)-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)benzamide (10t). Yield: 72%; 1H NMR (400 MHz, DMSO) δ 11.48 (s, 1H), 8.71 (d, J = 2.4 Hz, 1H), 8.34–7.91 (m, 2H), 7.58 (d, J = 8.4 Hz, 1H), 7.50 (s, 1H), 7.30 (s, 1H), 4.32 (d, J = 4.8 Hz, 2H), 3.83 (d, J = 9.6 Hz, 2H), 3.25 (t, J = 11.2 Hz, 2H), 3.15–3.06 (m, 2H), 3.03 (m, 1H), 2.75 (s, 2H), 2.38 (s, 2H), 2.27 (s, 3H), 2.10 (s, 3H), 1.66 (m, 6H), 1.57–1.44 (m, 2H), 0.82 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, DMSO) δ 168.71, 161.53, 150.10, 149.31, 149.06, 147.70, 140.37, 139.81, 137.75, 134.67, 133.92, 132.61, 124.25, 123.11, 120.96, 120.67, 111.52, 66.31, 57.78, 40.95, 34.58, 30.24, 26.61, 24.08, 22.13, 21.89, 15.93, 14.68, 12.70. ESI-MS m/z 549.2 (M + H)+.
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-(6-(4-methylpiperazin-1-yl)pyridin-3-yl)benzamide (10u). Yield: 82%; 1H NMR (400 MHz, DMSO) δ 11.48 (s, 1H), 8.39 (d, J = 2.4 Hz, 1H), 8.12 (t, J = 4.8 Hz, 1H), 7.75 (m, 1H), 7.36 (s, 1H), 7.17 (s, 1H), 6.89 (d, J = 8.8 Hz, 1H), 4.31 (d, J = 4.8 Hz, 2H), 3.83 (d, J = 10.2 Hz, 2H), 3.52 (s, 4H), 3.25 (t, J = 11.2 Hz, 2H), 3.08 (m, 2H), 3.00 (m, 1H), 2.71 (s, 2H), 2.41 (m, 8H), 2.23 (s, 3H), 2.11 (s, 3H), 1.64 (s, 7H), 1.51 (m, 2H), 0.82 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.10, 163.57, 158.36, 151.14, 149.59, 145.90, 140.75, 139.50, 135.86, 135.42, 132.93, 125.77, 123.08, 121.04, 119.94, 114.94, 106.84, 67.32, 58.38, 54.53, 45.80, 44.79, 41.52, 35.88, 30.45, 27.39, 24.85, 22.27, 22.19, 16.52, 14.74, 12.76. ESI-MS m/z 613.2 (M + H)+.
Enzyme inhibitory assays
The enzyme inhibitory assays were performed according to the AlphaLISA assay protocols (PerkinElmer Inc.).
Western blot analysis
After treatment with a series of concentrations of SKLB1049 for various days at 37 °C, cells were harvested, washed in ice-cold PBS, and lysed with RIPA buffer (Beyotime, China). And the protein concentrations were determined by the Bradford method. Proteins were separated by gel electrophoresis on 5–10% SDS-PAGE gels and probed with specific antibodies (Cell Signaling Technology, USA) including anti-H3K27me3, anti-H3K27me2, anti-EZH2, anti-H3 and anti-β-actin. All of the antibodies were used at a 1:1000 dilution, and the horseradish peroxidase-coupled secondary antibodies (Zhong Shan Golden Bridge Bio-technology, China) were used at 1:5000.
Cell viability assay
Human cancer cell lines used in this investigation were obtained from American Type Culture Collection (ATCC, Rockville, MD). The MTT assay was used to measure the cell viability after compounds treatment. Cells were seeded in 96-well microplates at a density of 2–5 × 103 cells per well, and then, cultured for 24 h. After treatment with various concentrations of compound for 4 or 6 days, 20 μL of MTT solution (5 mg mL−1) was added to each well and incubated 2–4 h at 37 °C, the formazan crystals were dissolved with 50 μL of acidified SDS (20%, w/v). The absorbance of each well was measured with Spectra MAX M5 microplate spectrophotometer at 570 nm wavelength, and the median inhibitory concentration (IC50) of each cell line was calculated.
Flow cytometry for cell cycle analysis
The cell cycle was measured by FCM assay. Approximately 1 × 105 cells were collected after treatment with vehicle or SKLB1049 in various concentrations for 14 days and fixed with 70% ethanol overnight. The cells were then washed with cold PBS and stained with 50 μg mL−1 propidium iodide containing 100 μg mL−1 RNase, and 0.1% Triton X-100. The cell cycle profiles were determined on a FACS Calibur flow cytometer (Becton Dicknson, USA) and analyzed using the ModFit LT 3.2 software (Verity Software House, USA).
Acknowledgements
We thank Zhejiang Apeloa Medical Technology Co., Ltd, for their technical and financial support. We also thank Lei Li of State Key Laboratory of Biotherapy (Sichuan University) for NMR measurements.
Notes and references
- R. Siegel, J. Ma, Z. Zou and A. Jemal, Ca-Cancer J. Clin., 2014, 64, 9–29 CrossRef PubMed.
- S. Wee, D. Dhanak, H. Li, S. A. Armstrong, R. A. Copeland, R. Sims, S. B. Baylin, X. S. Liu and L. Schweizer, Ann. N. Y. Acad. Sci., 2014, 1309, 30–36 CrossRef CAS PubMed.
- R. Margueron and D. Reinberg, Nature, 2011, 469, 343–349 CrossRef CAS PubMed.
- J. A. Simon and C. A. Lange, Mutat. Res., 2008, 647, 21–29 CrossRef CAS PubMed.
- F. M. Raaphorst, F. J. van Kemenade, T. Blokzijl, E. Fieret, K. M. Hamer, D. P. E. Satijn, A. P. Otte and C. J. L. M. Meijer, Am. J. Pathol., 2000, 157, 709–715 CrossRef CAS.
- H. P. J. Visser, M. J. Gunster, H. C. Kluin-Nelemans, E. M. M. Manders, F. M. Raaphorst, C. J. L. M. Meijer, R. Willemze and A. P. Otte, Br. J. Haematol., 2001, 112, 950–958 CrossRef CAS.
- O. Tamgue, C.-S. Chai, L. Hao, J.-C. D. Zambe, W.-W. Huang, B. Zhang, M. Lei and Y.-M. Wei, Asian Pac. J. Cancer Prev., 2013, 14, 5663–5669 CrossRef.
- C. G. Kleer, Q. Cao, S. Varambally, R. Shen, I. Ota, S. A. Tomlins, D. Ghosh, R. G. Sewalt, A. P. Otte, D. F. Hayes, M. S. Sabel, D. Livant, S. J. Weiss, M. A. Rubin and A. M. Chinnaiyan, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11606–11611 CrossRef CAS PubMed.
- N. Wagener, S. Macher-Goeppinger, M. Pritsch, J. Husing, K. Hoppe-Seyler, P. Schirmacher, J. Pfitzenmaier, A. Haferkamp, F. Hoppe-Seyler and M. Hohenfellner, BMC Cancer, 2010, 10, 524 CrossRef PubMed.
- E. Hodis, I. R. Watson, G. V. Kryukov, S. T. Arold, M. Imielinski, J. P. Theurillat, E. Nickerson, D. Auclair, L. Li, C. Place, D. Dicara, A. H. Ramos, M. S. Lawrence, K. Cibulskis, A. Sivachenko, D. Voet, G. Saksena, N. Stransky, R. C. Onofrio, W. Winckler, K. Ardlie, N. Wagle, J. Wargo, K. Chong, D. L. Morton, K. Stemke-Hale, G. Chen, M. Noble, M. Meyerson, J. E. Ladbury, M. A. Davies, J. E. Gershenwald, S. N. Wagner, D. S. Hoon, D. Schadendorf, E. S. Lander, S. B. Gabriel, G. Getz, L. A. Garraway and L. Chin, Cell, 2012, 150, 251–263 CrossRef CAS PubMed.
- Z. Li, Y. Wang, J. Qiu, Q. Li, C. Yuan, W. Zhang, D. Wang, J. Ye, H. Jiang, J. Yang and J. Cheng, OncoTargets Ther., 2013, 4, 2532–2549 Search PubMed.
- S. Varambally, S. M. Dhanasekaran, M. Zhou, T. R. Barrette, C. Kumar-Sinha, M. G. Sanda, D. Ghosh, K. J. Pienta, R. G. Sewalt, A. P. Otte, M. A. Rubin and A. M. Chinnaiyan, Nature, 2002, 419, 624–629 CrossRef CAS PubMed.
- S. Guo, J. K. Chan, J. Iqbal, T. McKeithan, K. Fu, B. Meng, Y. Pan, W. Cheuk, D. Luo, R. Wang, W. Zhang, T. C. Greiner and W. C. Chan, Clin. Cancer Res., 2014, 20, 3078–3086 CrossRef CAS PubMed.
- R. D. Morin, N. A. Johnson, T. M. Severson, A. J. Mungall, J. An, R. Goya, J. E. Paul, M. Boyle, B. W. Woolcock, F. Kuchenbauer, D. Yap, R. K. Humphries, O. L. Griffith, S. Shah, H. Zhu, M. Kimbara, P. Shashkin, J. F. Charlot, M. Tcherpakov, R. Corbett, A. Tam, R. Varhol, D. Smailus, M. Moksa, Y. Zhao, A. Delaney, H. Qian, I. Birol, J. Schein, R. Moore, R. Holt, D. E. Horsman, J. M. Connors, S. Jones, S. Aparicio, M. Hirst, R. D. Gascoyne and M. A. Marra, Nat. Genet., 2010, 42, 181–185 CrossRef CAS PubMed.
- R. D. Morin, M. Mendez-Lago, A. J. Mungall, R. Goya, K. L. Mungall, R. D. Corbett, N. A. Johnson, T. M. Severson, R. Chiu, M. Field, S. Jackman, M. Krzywinski, D. W. Scott, D. L. Trinh, J. Tamura-Wells, S. Li, M. R. Firme, S. Rogic, M. Griffith, S. Chan, O. Yakovenko, I. M. Meyer, E. Y. Zhao, D. Smailus, M. Moksa, S. Chittaranjan, L. Rimsza, A. Brooks-Wilson, J. J. Spinelli, S. Ben-Neriah, B. Meissner, B. Woolcock, M. Boyle, H. McDonald, A. Tam, Y. Zhao, A. Delaney, T. Zeng, K. Tse, Y. Butterfield, I. Birol, R. Holt, J. Schein, D. E. Horsman, R. Moore, S. J. Jones, J. M. Connors, M. Hirst, R. D. Gascoyne and M. A. Marra, Nature, 2011, 476, 298–303 CrossRef CAS PubMed.
- M. T. McCabe, H. M. Ott, G. Ganji, S. Korenchuk, C. Thompson, G. S. Van Aller, Y. Liu, A. P. Graves, A. D. P. Iii, E. Diaz, L. V. LaFrance, M. Mellinger, C. Duquenne, X. Tian, R. G. Kruger, C. F. McHugh, M. Brandt, W. H. Miller, D. Dhanak, S. K. Verma, P. J. Tummino and C. L. Creasy, Nature, 2012, 492, 108–112 CrossRef CAS PubMed.
- S. K. Knutson, S. Kawano, Y. Minoshima, N. M. Warholic, K. C. Huang, Y. Xiao, T. Kadowaki, M. Uesugi, G. Kuznetsov, N. Kumar, T. J. Wigle, C. R. Klaus, C. J. Allain, A. Raimondi, N. J. Waters, J. J. Smith, M. Porter-Scott, R. Chesworth, M. P. Moyer, R. A. Copeland, V. M. Richon, T. Uenaka, R. M. Pollock, K. W. Kuntz, A. Yokoi and H. Keilhack, Mol. Cancer Ther., 2014, 13, 842–854 CrossRef CAS PubMed.
- S. K. Knutson, N. M. Warholic, T. J. Wigle, C. R. Klaus, C. J. Allain, A. Raimondi, M. Porter Scott, R. Chesworth, M. P. Moyer, R. A. Copeland, V. M. Richon, R. M. Pollock, K. W. Kuntz and H. Keilhack, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7922–7927 CrossRef CAS PubMed.
- S. K. Knutson, T. J. Wigle, N. M. Warholic, C. J. Sneeringer, C. J. Allain, C. R. Klaus, J. D. Sacks, A. Raimondi, C. R. Majer, J. Song, M. P. Scott, L. Jin, J. J. Smith, E. J. Olhava, R. Chesworth, M. P. Moyer, V. M. Richon, R. A. Copeland, H. Keilhack, R. M. Pollock and K. W. Kuntz, Nat. Chem. Biol., 2012, 8, 890–896 CAS.
- W. Qi, H. Chan, L. Teng, L. Li, S. Chuai, R. Zhang, J. Zeng, M. Li, H. Fan, Y. Lin, J. Gu, O. Ardayfio, J. H. Zhang, X. Yan, J. Fang, Y. Mi, M. Zhang, T. Zhou, G. Feng, Z. Chen, G. Li, T. Yang, K. Zhao, X. Liu, Z. Yu, C. X. Lu, P. Atadja and E. Li, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 21360–21365 CrossRef CAS PubMed.
- K. D. Konze, A. Ma, F. Li, D. Barsyte-Lovejoy, T. Parton, C. J. MacNevin, F. Liu, C. Gao, X.-P. Huang, E. Kuznetsova, M. Rougie, A. Jiang, S. G. Pattenden, J. L. Norris, L. I. James, B. L. Roth, P. J. Brown, S. V. Frye, C. H. Arrowsmith, K. M. Hahn, G. G. Wang, M. Vedadi and J. Jin, ACS Chem. Biol., 2013, 8, 1324–1334 CrossRef CAS PubMed.
- S. Garapaty-Rao, C. Nasveschuk, A. Gagnon, E. Y. Chan, P. Sandy, J. Busby, S. Balasubramanian, R. Campbell, F. Zhao, L. Bergeron, J. E. Audia, B. K. Albrecht, J.-C. Harmange, R. Cummings and P. Trojer, Chem. Biol., 2013, 20, 1329–1339 CrossRef CAS PubMed.
- W. D. Bradley, S. Arora, J. Busby, S. Balasubramanian, V. S. Gehling, C. G. Nasveschuk, R. G. Vaswani, C. C. Yuan, C. Hatton, F. Zhao, K. E. Williamson, P. Iyer, J. Mendez, R. Campbell, N. Cantone, S. Garapaty-Rao, J. E. Audia, A. S. Cook, L. A. Dakin, B. K. Albrecht, J. C. Harmange, D. L. Daniels, R. T. Cummings, B. M. Bryant, E. Normant and P. Trojer, Chem. Biol., 2014, 21, 1463–1475 CrossRef CAS PubMed.
- T. Kaiho, K. San-Nohe, S. Kajiya, T. Suzuki, K. Otsuka, T. Ito, J. Kamiya and M. Maruyama, J. Med. Chem., 1989, 32, 351–357 CrossRef CAS.
- F. Kopp, C. F. Stratton, L. B. Akella and D. S. Tan, Nat. Chem. Biol., 2012, 8, 358–365 CrossRef CAS PubMed.
- N. Y. Wang, W. Q. Zuo, Y. Xu, C. Gao, X. X. Zeng, L. D. Zhang, X. Y. You, C. T. Peng, Y. Shen, S. Y. Yang, Y. Q. Wei and L. T. Yu, Bioorg. Med. Chem. Lett., 2014, 24, 1581–1588 CrossRef CAS PubMed.
- J. L. Burgess, N. Johnson, S. D. Knight, L. Lafrance, W. H. Miller, K. Newlander, S. Romeril, M. B. Rouse, X. Tian, S. K. Verma and D. Suarez, PCT Int. Appl., WO 2012005805, 2012.
- S. K. Verma, X. Tian, L. V. LaFrance, C. Duquenne, D. P. Suarez, K. A. Newlander, S. P. Romeril, J. L. Burgess, S. W. Grant, J. A. Brackley, A. P. Graves, D. A. Scherzer, A. Shu, C. Thompson, H. M. Ott, G. S. Aller, C. A. Machutta, E. Diaz, Y. Jiang, N. W. Johnson, S. D. Knight, R. G. Kruger, M. T. McCabe, D. Dhanak, P. J. Tummino, C. L. Creasy and W. H. Miller, ACS Med. Chem. Lett., 2012, 3, 1091–1096 CrossRef CAS PubMed.
- J. R. Simard, M. Plant, R. Emkey and V. Yu, Assay Drug Dev. Technol., 2013, 11, 152–162 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.