
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot conversion of cellobiose to mannose using a hybrid phosphotungstic acid–cerium oxide catalyst†
Zane C.
Gernhart
a,
Anuja
Bhalkikar
a,
John J.
Burke
a,
Kate O.
Sonnenfeld
a,
Chris M.
Marin
a,
Richard
Zbasnik
b and
Chin Li
Cheung
*a
aDepartment of Chemistry, University of Nebraska-Lincoln, Lincoln, NE 68588, USA. E-mail: ccheung2@unl.edu
bDepartment of Food Science and Technology, University of Nebraska-Lincoln, Lincoln, NE 68583, USA
First published on 17th March 2015
Abstract
A hybrid catalyst composed of phosphotungstic acid coated cerium oxide nanoparticles was demonstrated to catalyze the one-pot conversion of cellobiose, the disaccharide unit of cellulose, to a monosaccharide mixture of glucose and mannose. A high % conversion of cellobiose (up to 99%) was achieved resulting in a yield of mannose up to 15.8%. The yield of mannose from a glucose starting material was 22.8%, exceeding those of previous cerium-based glucose epimerization catalysts. The components of the hybrid material were revealed to function synergistically via a two-step process. Cellobiose was hypothesized to be first hydrolyzed to glucose, which was subsequently epimerized to mannose by the cerium ions leached from the catalyst. The 13C NMR spectroscopic study suggested that the epimerization likely occurred by way of a 1,2-carbon shift reaction mechanism.
1. Introduction
The negative environmental and economic impacts of using petroleum sources to produce fuels and chemicals necessitate the development of viable alternative feedstocks.1,2 The use of plant-based food stocks for this purpose has been widely accepted, but concerns have been raised about the economic impacts of using food for fuel and chemical production.3 Cellulose, a non-edible polymer which serves as a structural framework for plants, is one of the most abundant bio-renewable materials on earth and is a promising alternative to petroleum feedstocks.4 The cellulose polymer is composed of glucose monomer units linked by β-(1,4)-glycosidic bonds. Enzymatic depolymerization of cellulose into glucose units is already being utilized for commercial biofuels production.5,6 However, the production of other monosaccharides from cellulose sources has been investigated to a much lesser extent. Many of these sugars are valuable on their own and they have the potential to be converted into other high value products.7,8 For example, mannose, a monosaccharide which is only found as part of complex polysaccharides in nature, has many important uses such as a precursor for sweeteners, vitamin production, antitumor agents and immunoregulatory agents.9,10Research on the enzymatic production of mannose has been primarily focused on the conversion of fructose to mannose.9 While fructose is found in abundance in the edible portion of many plants, it is present in only small amounts in the non-edible biomass portion.11 The enzymatic production of mannose from glucose, a major building block of non-edible biomass, appears to be much less common. However, the successful use of inorganic catalysts for this transformation has been demonstrated through the Lobry de Bruyn–van Ekenstein reaction, Bilik reaction, and similar 1,2-carbon shift reactions.8,12 The possibility of combining an inorganic epimerization catalyst with a catalyst capable of causing cellulose depolymerization raises the exciting prospect that a one-pot hybrid catalyst system can be designed for the conversion of cellulose to mannose.
The first step in any one-pot catalytic conversion of cellulose to mannose is the depolymerization of cellulose to glucose. Solid acid catalysts are potential alternatives for activating cellulose hydrolysis because of their capability to function in a large range of temperatures and their general ease of separation from products.13–16 Examples of solid acid catalysts reported for cellulose processing include metal oxides, sulfonated carbon-based particles, polymer-based acids, zeolites, supported inorganic catalysts, and polyoxometalates (POMs).17 Of these catalysts, POMs are particularly appealing due to their highly acidic nature and low corrosiveness.18 For example, phosphotungstic acid (PWA, H3PW12O40) has been demonstrated to hydrolyze cellulose with yields of glucose greater than 50% and glucose selectivity greater than 90%.19 Catalysts composed of POMs supported on or constructed in hybrid with a variety of metal oxide particles have also been previously reported,20,21 with some of the earliest work focusing on the PWA/silica system.22 POM supported on silica was demonstrated to effectively depolymerize cellulose in the conversion of carbohydrate-rich weeds to 5-hydroxymethylfurfural.21 However, the production of mannose was not observed or expected for this reaction because of the lack of an epimerization catalyst.
An essential second step in a one-pot catalytic conversion of cellulose to mannose is the epimerization of glucose to mannose. Only a handful of inorganic catalyst systems have been reported for this epimerization. Typical examples include homogeneous catalysts such as metal ions complexed with amines12,23 and solid acid catalysts such as tin beta (Sn-beta) zeolites.24,25 The use of Sn-beta zeolites with sodium tetraborate salts has proven quite successful for the epimerization of glucose with yields of mannose reaching as high as 21% when the glucose to sodium tetraborate reactant molar ratio was 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.24 Lanthanide ions, including cerium, have also been demonstrated to effectively catalyze the epimerization of glucose to mannose via a 1,2-carbon shift.12,23 The use of Ce3+ in the presence of diamines was shown to effectively epimerize glucose with yields of mannose as high as 14%.23 Previous research has suggested that understanding homogeneous catalyst systems can allow for the design of improved solid catalysts.26
:1.24 Lanthanide ions, including cerium, have also been demonstrated to effectively catalyze the epimerization of glucose to mannose via a 1,2-carbon shift.12,23 The use of Ce3+ in the presence of diamines was shown to effectively epimerize glucose with yields of mannose as high as 14%.23 Previous research has suggested that understanding homogeneous catalyst systems can allow for the design of improved solid catalysts.26
Herein we report the effectiveness of a hybrid catalyst composed of PWA coated on cerium oxide nanoparticles (hereafter referred to as PWA/CeO2−x) towards the conversion of cellobiose into glucose and mannose. CeO2−x, 0 < x ≤ 0.5, is a widely used support material which has previously been used to prepare cerium oxide supported PWA. This material was demonstrated to catalyze the oxidation of >99% of sulfur compounds from model oil systems with high recyclability.20 The presence of Ce3+ and Ce4+ valence sites on the surface of nanostructured CeO2−x (ref. 27) in conjunction with its demonstrated ability to support PWA led to the hypothesis that it would be an effective catalyst for the production of mannose from cellulose. Cellobiose was used to evaluate this catalyst system because it is a dimer of glucose molecules connected by a β-(1,4)-glycosidic bond. Its molecular similarity to cellulose, which is a polymer of glucose monomer units connected by β-(1,4)-glycosidic bonds, has allowed it to be used as a model cellulose system.28–31 Our investigations of the roles of each catalyst component revealed that the PWA/CeO2−x catalyst demonstrates a synergistic ability to convert cellobiose to monosaccharides. This synergistic effect was attributed to the epimerization of glucose to mannose. 13C NMR was utilized to investigate the mechanism behind this conversion. Catalyst leaching was investigated to determine possible roles of leached cerium ions from the catalyst in the conversion of cellobiose to monosaccharides.
2. Experimental methods
2.1 Preparation of catalysts
Phosphotungstic acid decorated cerium oxide nanoparticles (7 mol% PWA/CeO2−x) were synthesized using a previously reported co-precipitation method.20 In a typical synthesis, 2.32 g of cerium(IV) ammonium nitrate ((NH4)2Ce(NO3)6, 99.5%, Alfa-Aesar, Ward Hill, MA) were dissolved in a solution containing 0.97 mL of acetic acid, 10 mL of ethanol, and 1 mL of 18 MΩ cm deionized water. The resulting solution (Solution A) was stirred at room temperature for 1 h. Solution B was made by dissolving 0.994 g of dodecatungstophosphoric acid hydrate (H3O40PW12·xH2O, reagent grade, Sigma-Aldrich, St. Louis, MO) in a solution of 10 mL of ethanol and 1 mL of 18 MΩ cm deionized water. Solution B was added drop-wise via a glass burette to Solution A while being stirred. The resulting mixture was left stirring overnight, followed by aging at 40 °C for 6 h. and then 100 °C for 2 h. The obtained solid was transferred into a crucible and calcined at 400 °C for 4 h. Cerium oxide (CeO2−x) nanoparticles which were used as a control comparison to the PWA/CeO2−x particles were synthesized using a similar precipitation method but without the addition of Solution B.2.2 Materials characterization
The crystal structure of the as-synthesized samples was investigated using powder X-ray diffraction (XRD) (PANalytical Empyrean, Westborough, MA). The average wavelength of the Cu Kα X-ray source was 1.544 Å. Diffraction peak assignments corresponding to fluorite-structured cerium(IV) oxide (CeO2) in the XRD data were indexed using the ICDD data card #04-013-4361. Further analysis of the crystal structures and chemical properties of the samples were made using high resolution transmission electron microscopy (HRTEM) operated at 200 kV and energy-dispersive X-ray spectroscopy (EDX) (Tecnai F-20, FEI, Hillsboro, OR).2.3 Activity evaluation of catalysts
The catalytic activity of 7 mol% PWA/CeO2−x towards the conversion of cellobiose into glucose and mannose was investigated using a MARS 6 Microwave Reaction System (CEM, Matthews, NC). In a typical reaction, 0.021 g of 7 mol% PWA/CeO2−x was transferred into a GlassChem reaction vessel (CEM, Matthews, NC). 10 mL of a 50 mM solution of D-(+)-cellobiose (C12H22O11, 98+%, Alfa-Aesar, Ward Hill, MA) was then added to the reaction vessel. Water served as the solvent for all reactions reported in this study. The vessel was sealed and placed inside the microwave where it was heated to the pre-determined reaction temperature over a 10 min-period. The reaction mixture was stirred using a Teflon stir bar and the reaction temperature was monitored using a fiber optic probe. The reaction temperatures evaluated were 140 °C, 160 °C, and 180 °C. The reaction pressure was estimated using an EasyPrep autoclave vessel (CEM, Matthews, NC) with reaction volume scaled to match those carried out previously. The pressures were found to be 225, 446, and 784 kPa at 140 °C, 160 °C, and 180 °C, respectively. The reaction mixture was held at the pre-determined temperature for 30, 60, 90, or 120 minutes, after which it was then allowed to cool to room temperature. A 1.5 mL product sample was centrifuged and syringe filtered with a 0.2 μm pore-size polytetrafluoroethylene membrane (VWR International, Radnor, PA) before its analysis. To investigate the effect of each catalyst component on the reaction, the foregoing procedure was repeated with cerium oxide nanoparticles (CeO2−x) and PWA hydrate of masses 0.0093 g and 0.0127 g, respectively. These masses were chosen so that they were equal to the maximum weight amounts of each catalyst component which could have been present in 0.021 g of the 7 mol% PWA/CeO2−x catalyst. In order to determine the effect of microwave heating, a series of reactions was performed in a convection oven at 160 °C using Teflon-lined stainless steel autoclaves (Parr, Moline, IL). The maximum % conversion of cellobiose and yields of mannose were similar to those obtained using microwave heating but required longer reaction times. These results are provided for comparison in the ESI.†2.4 Reaction product analysis by HPLC
The saccharide content in the reaction products was analyzed for each trial in our study using a method developed by the National Renewable Energy Laboratory.32 Briefly, a 20 μL product sample solution was injected into a high performance liquid chromatography (HPLC) instrument (Waters, Milford, MA) with a 0.005 M sulfuric acid (H2SO4, 99.999%, Sigma-Aldrich, St. Louis, MO) mobile phase flowing at 0.6 mL min−1. An Aminex HPX-87H column (Bio-Rad Laboratories Inc., Hercules, CA) heated at 55 °C was used to separate the product sample solution into different saccharide components. The separated saccharide components were detected using a Waters 410 Differential Refractometer (Waters, Milford, MA) heated at 35 °C. Confirmation of mannose in the sample was made using a Dionex ICS-3000 HPLC (ThermoScientific, Sunnyvale, CA) equipped with a Dionex Carbopac PA20 column (ThermoScientific, Sunnyvale, CA). A second confirmation was made using an Aminex HPX-87P column (Bio-Rad Laboratories Inc., Hercules, CA).The concentrations of cellobiose, glucose and mannose in the product samples were determined by integrating the areas of corresponding refractive index peaks and comparing these areas with those of the respective standard. Percentage (%) conversion of cellobiose, % product yields for glucose and mannose, and % product selectivity for each monosaccharide were reported according to eqn (1)–(3) below:
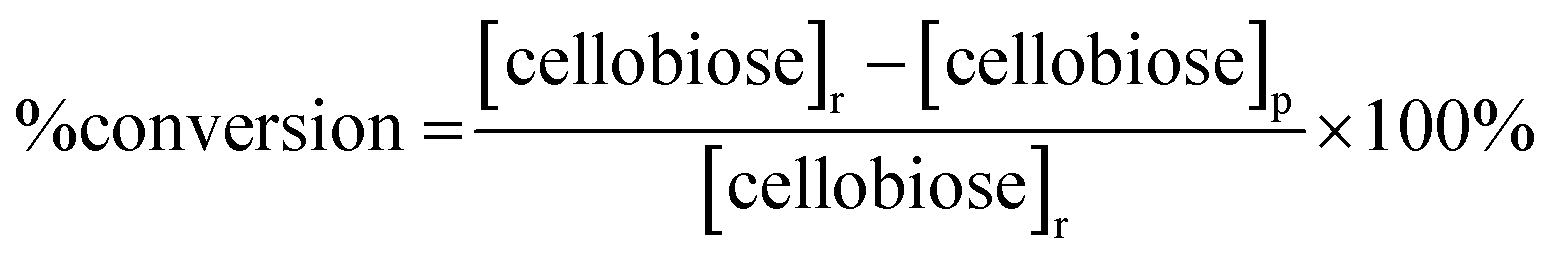 | (1) |
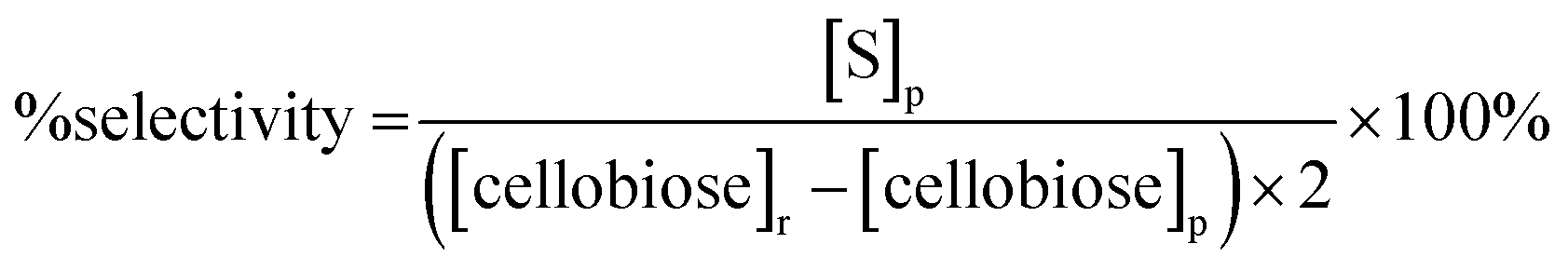 | (2) |
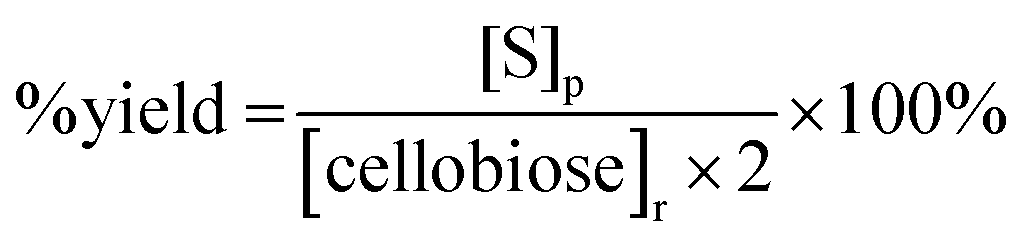 | (3) |
2.5 Saccharide epimerization study
To evaluate the effect of the hybrid catalyst and individual components on glucose epimerization, catalysis experiments at 160 °C were repeated with a 25 mM solution of D-(+)-glucose (C6H12O6, ≥99.5%, Sigma-Aldrich, St. Louis, MO). In a typical reaction, 0.021 g of 7 mol% PWA/CeO2−x was transferred into a GlassChem reaction vessel (CEM, Matthews, NC). 10 mL of a 25 mM solution of D-(+)-glucose was then added to the reaction vessel. The vessel was sealed and placed inside the microwave where it was heated to 160 °C over a 10 min-period. A standard 30 min reaction duration was used for all trials. The reaction mixture was stirred using a Teflon stir bar and the reaction temperature was monitored using a fiber optic probe. A 1.5 mL product sample was centrifuged and syringe filtered with a 0.2 μm pore-size polytetrafluoroethylene membrane before its analysis by HPLC. To investigate the effect of each catalyst component on the reaction, the foregoing procedure was repeated with cerium oxide nanoparticles (CeO2−x) and PWA hydrate of masses 0.0093 g and 0.0127 g, respectively.To investigate the conversion of glucose to mannose by the PWA/CeO2−x catalyst, a 5 mg mL−1 solution of glucose with a 13C label at the C1 position (D-glucose, 1-13C, 98–99%, Cambridge Isotope Laboratories, Inc., Andover, MA), was prepared with 18 MΩ cm deionized water. 10 mL of this solution was added to a GlassChem reaction vessel containing the same weight amount of catalyst as described in the above cellobiose conversion study. The solution underwent the standard 30 min microwave-heated reaction at 160 °C. Both the starting solution and reaction products were analyzed by 13C NMR using a Bruker Avance III-HD 400 MHz Spectrometer (Bruker BioSpin, Billerica, MA). One drop of deuterated water (D2O) was added to each NMR sample. Thirty two scans of standard 13C {1H} NMR with a pulse width of 30° were performed to obtain each NMR spectrum.
2.6 Catalyst leaching study
Metal ion leaching has been demonstrated to have a large impact on the catalytic activity of many reported catalyst systems.33 To investigate the presence and role of leaching during the course of our reaction, a 25 mM solution of D-(+)-glucose (C6H12O6, ≥99.5%, Sigma-Aldrich, St. Louis, MO) was heated to 160 °C in the presence of 7 mol% PWA/CeO2−x. Once the reaction mixture reached 160 °C, the heating was stopped and the reaction vessel was allowed to cool to room temperature. To remove the catalyst particles, the product mixtures were centrifuged and filtered using a 0.2 μm polytetrafluoroethylene filter. The filtered solution was frozen at −14 °C. After thawing, the solution was centrifuged and filtered using a 0.02 μm Whatman Anotop filter (GE Healthcare Bio-Sciences, Pittsburgh, PA). A 1.0 mL sample of filtered solution was removed for analysis by HPLC. It was then placed back in the microwave and heated at 160 °C for an additional 30 minutes. The reaction products after the second microwave heating were analyzed by HPLC to determine the effect of filtration on the conversion of cellobiose and by inductively coupled plasma mass spectrometry (ICP-MS, ThermoScientific, Waltham, MA) to determine the concentration of cerium in the catalyst products.3. Results and discussion
3.1 Structural and chemical characterization of the catalysts
Both powder XRD patterns of the CeO2−x and PWA/CeO2−x demonstrate the presence of a fluorite-phase (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) cerium oxide (Fig. 1). The XRD pattern of the 7 mol% PWA/CeO2−x samples calcined at 400 °C is consistent with a previously reported pattern.20 The presence of tungsten oxide (WO3) is indicated by a peak at a 2 theta of 24.39°. These features in the XRD pattern are consistent with the partial decomposition of PWA during the calcination process.20,34
m) cerium oxide (Fig. 1). The XRD pattern of the 7 mol% PWA/CeO2−x samples calcined at 400 °C is consistent with a previously reported pattern.20 The presence of tungsten oxide (WO3) is indicated by a peak at a 2 theta of 24.39°. These features in the XRD pattern are consistent with the partial decomposition of PWA during the calcination process.20,34
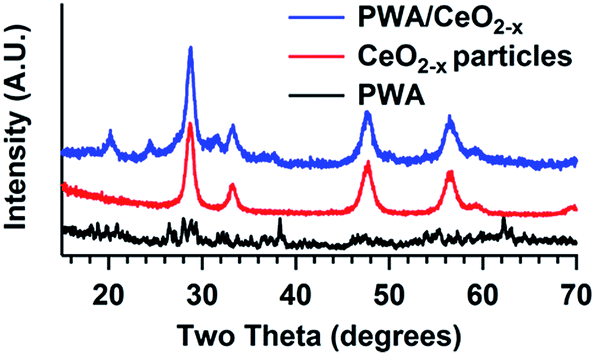 | ||
| Fig. 1 XRD of (top) PWA/CeO2−x, (middle) CeO2−x nanoparticles, and (bottom) PWA. | ||
HRTEM images of the CeO2−x and PWA/CeO2−x samples also reveal a fluorite-structured cerium oxide crystal lattice for both materials. The HRTEM image of a PWA/CeO2−x sample in Fig. 2a displays lattice spacing of 3.24 Å, which corresponds to the distance between the (−111) and (1−11) planes of fluorite-structured cerium dioxide when viewed down the [110] direction. An observed 3% increase in lattice spacing for the PWA/CeO2−x could be explained by possible increased lattice strain in this hybrid material. The HRTEM image of the CeO2−x sample shows characteristic lattice spacings for cerium dioxide (Fig. 3a). The identified lattice spacing of 3.15 Å corresponds to the distance between adjacent (111) planes. Another set of identified lattice spacings of 1.95 Å corresponds to one half of the distance between (011) planes. This matches well with the location of a second plane of cerium atoms.
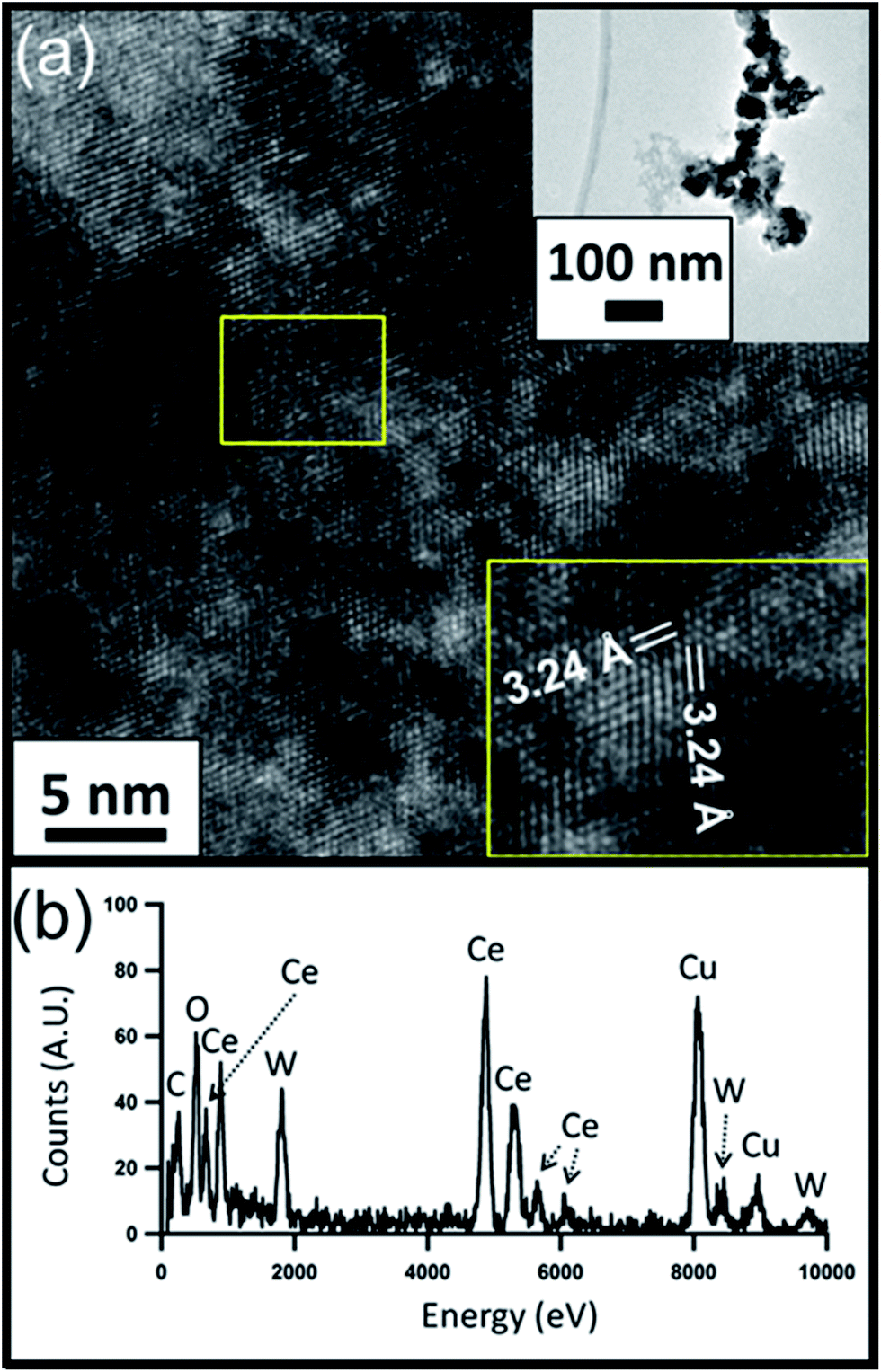 | ||
| Fig. 2 (a) HRTEM image of the 7 mol% PWA/CeO2−x catalyst with zoomed out image (top inset) and measured lattice spacing (bottom inset); (b) corresponding EDX spectrum. | ||
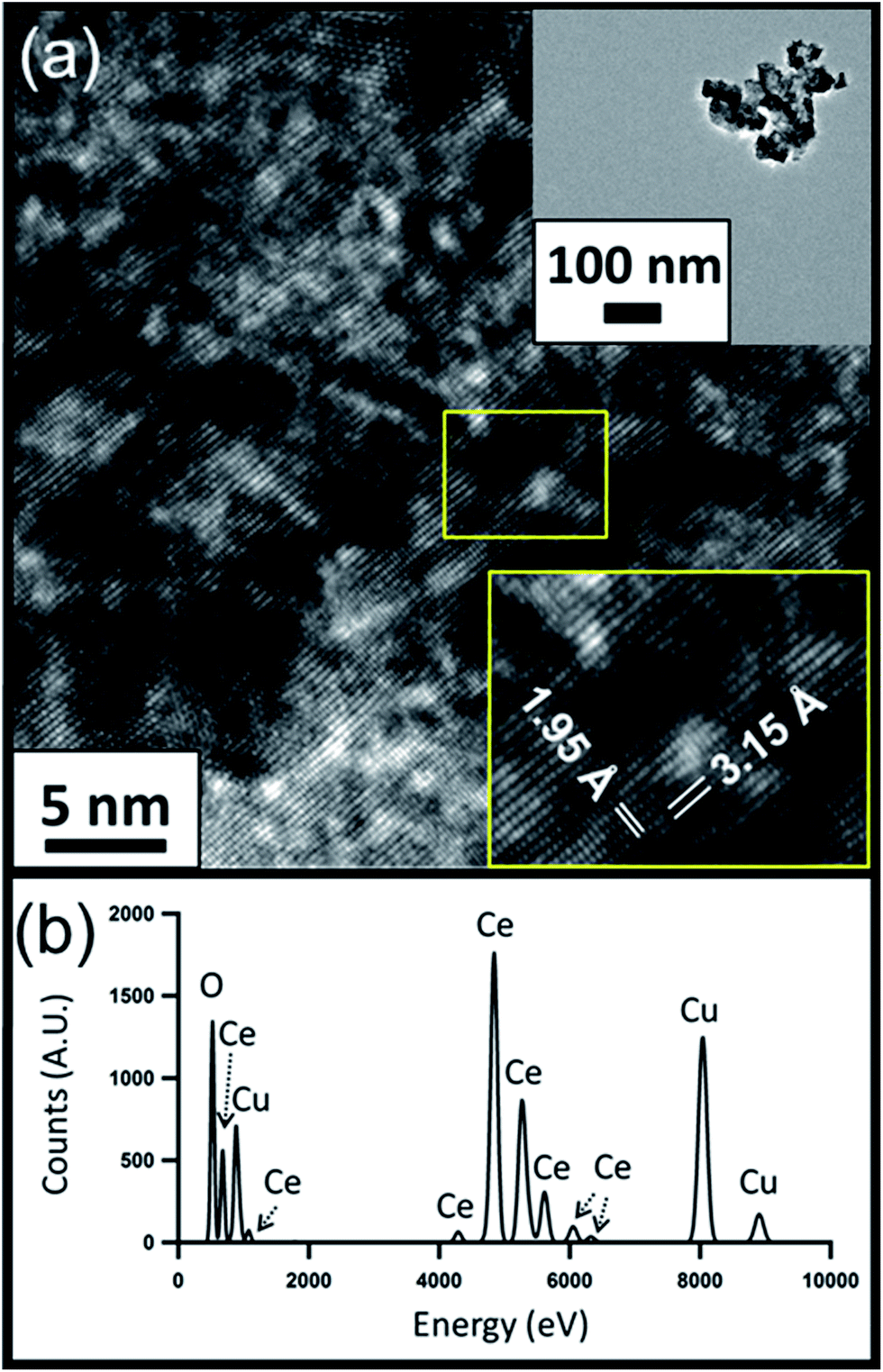 | ||
| Fig. 3 (a) HRTEM image of CeO2−x nanoparticle clusters with zoomed out image (top inset) and measured lattice spacing (bottom inset); (b) corresponding EDX spectrum. | ||
Elemental analysis of the PWA/CeO2−x by EDX confirmed the presence of both cerium and tungsten in the PWA/CeO2−x samples (Fig. 2b). In contrast, no peaks indicating the presence of tungsten were observed in the EDX data of the CeO2−x samples, in spite of a longer integration time being allowed for the CeO2−x sample than for the PWA/CeO2−x sample (Fig. 3b).
3.2 Effects of reaction temperature and duration on cellobiose conversion
The effects of reaction temperature and duration on the catalytic cellobiose conversion were investigated using a range of microwave heating temperatures (Fig. 4) and reaction periods (Fig. 5). The primary reaction products were identified by HPLC as glucose and mannose. For reactions performed with durations of 30 min, when the reaction temperature was increased from 140 °C to 180 °C, the conversion of cellobiose increased from 15.8% to 99.1%. However, the resulting product selectivity for glucose decreased from 75.1% to 27.7%. The % yield of glucose was observed to reach a maximum value of 32.6% at 160 °C and decrease at higher reaction temperatures. The % yield of mannose was observed to increase with an increase in reaction temperature over the evaluated range, and it reached 15.4% at a reaction temperature of 180 °C.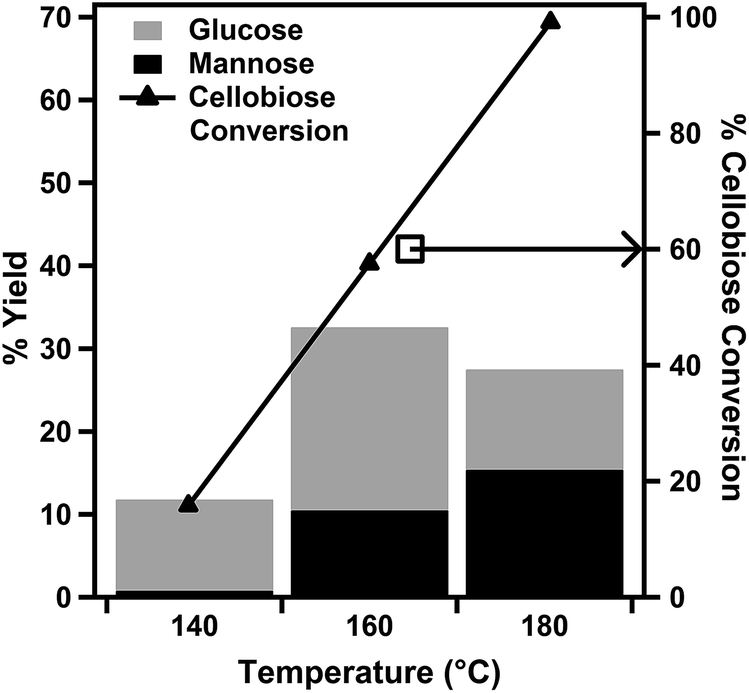 | ||
| Fig. 4 Effect of reaction temperature on 30 minute cellobiose hydrolysis reactions with the 7 mol% PWA/CeO2−x catalyst. | ||
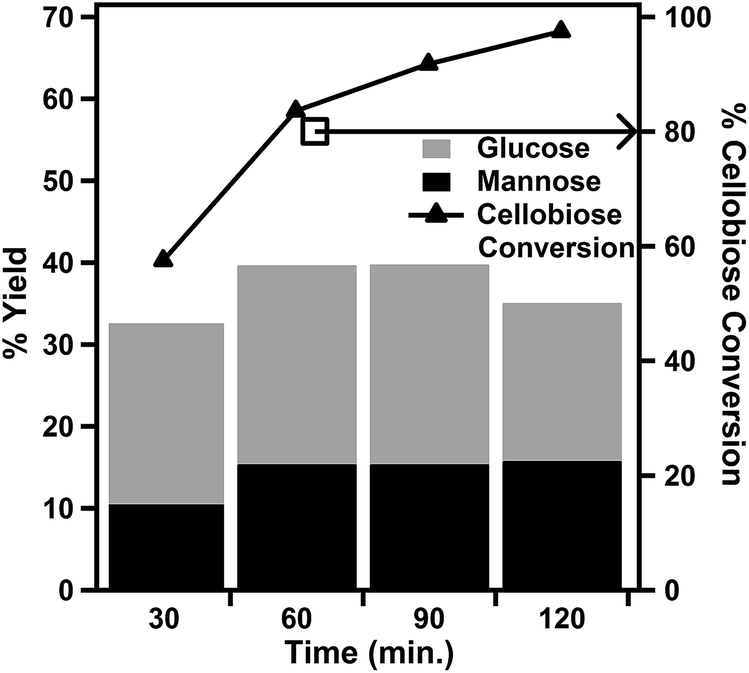 | ||
| Fig. 5 Effect of reaction duration on cellobiose hydrolysis reactions at 160 °C using the 7 mol% PWA/CeO2−x catalyst. | ||
The effect of reaction durations from 30 minutes to 2 hours was investigated at a fixed reaction temperature of 160 °C (Fig. 5). The conversion of cellobiose was found to increase with an increase in reaction duration. The % yield of glucose was observed to increase as the reaction time was increased from 30 minutes to 1 hour. However, there was little change to the % yield of glucose for reaction durations from 1 hour to 90 minutes. As the reaction duration was increased to 2 hours, the % yield of glucose was observed to decrease. The only evaluated reaction duration within the evaluated temperature range where both the % conversion of cellobiose and % yield of glucose were above 50% was at 30 minutes.
The production of mannose was also influenced by reaction duration. The % yield of mannose followed a similar trend as that of glucose up to 90 minutes. After a 90 min reaction, while the yield of glucose began to fall, the yield of mannose began to increase slightly. The highest yield of mannose in our reaction duration study was 15.8%. This was the result of a 120 min reaction at 160 °C. This % yield of mannose was similar to the 14% yield previously reported for a homogeneous cerium catalyst. However, while the previously reported catalyst system utilized glucose as the starting reactant, our catalyst was able to achieve similar yields with a cellooligosaccharide chemical in a one-pot reaction.23 This yield is also similar to those achieved using 1% Ru–5% Ni/mesoporous carbon (MC) and 1% Ir–5% Ni/MC catalysts with microcrystalline cellulose.35 However, it should be noted that these reactions were performed at 245 °C in the presence of 6 MPa of hydrogen. A noteworthy advantage of this 7 mol% PWA/CeO2−x catalyst is its ability to produce mannose under acidic conditions which are more similar to those used for cellulose processing than the basic conditions which were previously reported.12,36
3.3 Evaluation of the catalytic activity of individual catalyst components on cellobiose conversion
To determine the effect of each component of the PWA/CeO2−x catalyst system on the cellobiose conversion reaction, a series of control experiments were performed (Table 1). The effects of heating a cellobiose solution without any catalyst and with each catalyst component added separately were compared to the effects of heating a cellobiose solution with 7 mol% PWA/CeO2−x. When the 50 mM cellobiose solution was heated at 160 °C for 30 minutes without any catalyst in the solution, the obtained conversion of cellobiose was 8.9%. When an amount of CeO2−x equal to the amount found in 0.021 g of 7 mol% PWA/CeO2−x was added to a 50 mM cellobiose solution, the corresponding conversion of cellobiose was determined to be 9.0%. It is worth noting that the presence of CeO2−x by itself appears to have little effect on the conversion of cellobiose. When an amount of PWA equal to the amount in 0.021 g of 7 mol% PWA/CeO2−x was added to a 50 mM cellobiose solution, a 47.1% conversion was obtained.| Catalyst | Catalyst mass (g) | Conversion (%) | Glucose yield (%) | Glucose selectivity (%) | Mannose yield (%) | Mannose selectivity (%) |
|---|---|---|---|---|---|---|
| 7 mol% PWA/CeO2−x | 0.0210 | 57.5 | 32.6 | 56.7 | 10.5 | 18.2 |
| PWA hydrate | 0.0127 | 47.1 | 37.6 | 79.9 | 0.8 | 1.7 |
| CeO2−x | 0.0093 | 9.0 | 4.5 | 51.1 | 0.5 | 5.2 |
| No catalyst | — | 8.9 | 4.6 | 51.8 | 0.0 | 0.0 |
A zeroth-order approximation was made that a base conversion of 8.9% was achieved just by heating the solution. By subtracting this base conversion from the total percent conversion of a reaction with one catalyst component, the resulting number can be designated as the % conversion caused by that component (eqn (4) and (5)). Likewise, the % conversion attributed to the hybrid catalyst system can be obtained when the base conversion achieved by heating is subtracted from results produced by the 7 mol% PWA/CeO2−x catalyst (eqn (6))
| %conversion by CeO2−x = %CCeO2−x − %CHeating | (4) |
| %conversion by PWA = %CPWA − %CHeating | (5) |
| %conversion by PWA/CeO2−x = %CPWA/CeO2−x − %CHeating | (6) |
The effects of a hybrid catalyst of PWA and CeO2−x on the conversion yield were compared to that of a reaction containing the two catalyst components added separately in the same solution. For the reaction with separate catalyst components not in hybrid form, it was found that the % conversion of cellobiose was 56.7%, the yield of glucose was 37.6% and the yield of mannose was 7.9%. Hence, the effect of having the two components in a hybrid form appears negligible in terms of % cellobiose conversion. However, a decreased yield of glucose for the hybrid material and slightly higher yield of mannose suggest that the hybrid material could have a positive effect on this step of the conversion.
3.4 Epimerization of glucose to mannose
In the preceding sections, it was noted that the % yield of mannose increased as that of glucose decreased. This observation led to the hypothesis that glucose was being converted to mannose after the hydrolysis of cellobiose. To verify this hypothesis, a 25 mM glucose solution mixed with the 7 mol% PWA/CeO2−x catalyst was heated at 160 °C for 30 minutes. All other reaction conditions were identical to those used for the conversion of cellobiose. Under these conditions, a 54.3% conversion of glucose and a 22.8% yield of mannose were obtained (Table 2). When a glucose solution with no catalyst was heated to 160 °C and held at this reaction temperature for 30 min, the resulting conversion was 11.0% (variable CHeating in eqn (4)–(6)). Almost no change was observed between the conversion for a glucose solution heated without catalyst and that for a glucose solution heated with CeO2−x. The conversion caused by an amount of PWA equal to the molar amount which could be present in 7 mol% PWA/CeO2−x was 12.2%. When the 7 mol% PWA/CeO2−x was used as a catalyst, the conversion was observed to increase to 54.3%. However, when the conversions resulting from each component are independently assessed to account for the effect of heating (using eqn (4) and (5)), the sum of the components is ca. 42% less than the conversion caused by 7 mol% PWA/CeO2−x (eqn (6)).| Catalyst | Catalyst mass (g) | Conversion (%) | Mannose yield (%) | Mannose selectivity (%) |
|---|---|---|---|---|
| 7 mol% PWA/CeO2−x | 0.0210 | 54.3 | 22.8 | 42.1 |
| PWA hydrate | 0.0127 | 12.2 | 2.4 | 19.9 |
| CeO2−x | 0.0093 | 10.3 | 0.9 | 9.2 |
| No catalyst | — | 11.0 | 4.4 | 39.7 |
The 22.8% yield of mannose achieved using this hybrid catalyst with glucose is higher than the 14% yield previously reported for reactions catalyzed using cerium ions with amines instead.23 This yield is comparable to the 21% yield achieved with recently reported high-performance Sn-beta epimerization catalysts in the presence of sodium tetraborate.24 A possible explanation for this increase in yield when compared to the previous use of cerium ion catalyst is that the acidic reaction conditions minimized the formation of undesirable 2-ketoses through the Lobry de Bruyn–van Ekenstein reaction, which commonly proceeds under basic conditions.12
The synergistic effect observed in the conversion of cellobiose using the PWA/CeO2−x catalyst system is likely due to the conversion of glucose produced from the hydrolysis of cellobiose into mannose. As the glucose was removed from the reaction mixture, through conversion to mannose, the conversion of cellobiose to glucose increased. The epimerization of glucose to mannose catalyzed by cerium ions has previously been reported.23 However, to the best of our knowledge, no reports have been made of this epimerization in the presence of CeO2−x. This raises the possibility that the synergistic effect observed in the catalyst system is a result of homogeneous catalytic reactions involving leached cerium ions from the hybrid catalyst. Due to the important role of this effect in other catalyst systems which were once thought to be entirely heterogeneous,37,38 it is critical that this effect be investigated for PWA/CeO2−x.
3.5 Investigation of catalyst leaching
Understanding the effect of leaching in this catalyst system is a critical step towards utilizing and improving supported PWA catalyst design. In the past, leaching was often considered detrimental.39 However, recently, researchers have begun to view leaching as another tool to be harnessed in designing new catalyst systems.33 This effect has been studied extensively for platinum metal catalysts which are often used in organic synthesis. The activity of these catalysts depends on the form and chemical identities of ions leached from the metal particles into the solution.38 Understanding this relationship has allowed researchers to make modifications to the reaction mixture or catalyst system to better control reaction products.33In order to investigate the effect of catalyst leaching on mannose production from glucose, the catalyst material was filtered from the reaction once the mixture had reached 160 °C. The change in the yield of mannose was then monitored for the remainder of the reaction. It was expected that if no leaching had occurred, little to no change in the yield of mannose would be observed after filtering. The percent conversion of glucose after heating to 160 °C and cooling was 14.8% and the yield of mannose was 8.6%. After the reaction mixture was filtered to remove catalyst nanoparticles, it was placed back in the microwave for a further 30 minutes of reaction at 160 °C. The resulting percent conversion of glucose increased to 56.2% and the yield of mannose increased to 22.2%. This conversion is remarkably close to the conversion achieved in the presence of the hybrid catalyst system without any filtration treatments (Table 2). The concentration of cerium in the reactants after filtering was measured to be ca. 50 ppm by ICP-MS.
The recyclability of the PWA/CeO2−x catalyst was studied to further investigate the role of leaching in the catalyst system. After a standard 30 minute reaction with cellobiose at 160 °C, the catalyst was collected by centrifugation and rinsed with water. The catalyst was then placed back in a reaction vessel with a new cellobiose solution and heated for another 30 minutes at 160 °C. The % conversion of cellobiose with a recycled catalyst was 23.3% and the % glucose yield was 15.5%. Neither value reached 50% of the original value achieved before recycling (Table 1). However, the value of mannose yield showed the greatest relative change with a yield of only 0.67% after recycling compared to a yield of 10.5% before. The decreased yields from the recycled catalyst were attributed to the leaching of cerium ions and PWA from the catalyst. The recycled catalyst likely did not contain enough acidic materials to cause the leaching of the cerium ions, which are required for epimerization to occur. This hypothesis was further evaluated by adding a mass of PWA equal to that in the original catalyst to the recycled catalyst material. After repeating the reaction under the same conditions, it was found that the yield of mannose produced reached 83% of the yield before recycling with additional PWA.
Previous studies on epimerization using cerium ions suggest that the conversion of glucose to mannose occurs via a 1,2-carbon shift.23 A recent study of Sn-beta zeolites has demonstrated a reliable method to investigate this 1,2-carbon shift using 13C NMR and D-(1-13C) glucose as the starting chemical.24 To determine whether the same mechanism was responsible for the conversion using our 7 mol% PWA/CeO2−x catalyst, we applied the catalyst to convert a D-(1-13C) glucose solution at 160 °C for 30 minutes and performed 13C NMR experiments to analyze the reaction products. The NMR spectrum of our initial D-(1-13C) glucose solution contained peaks at δ = 97.0 and 93.2 ppm, which correspond to the 13C at the C1 position of the β-pyranose and α-pyranose forms of glucose. Their locations are consistent with NMR features in previous reports for D-(1-13C) glucose24 (Fig. 6a). After the standard microwave reaction, a new additional set of peaks at δ = 71.8 and 72.4 ppm was observed (Fig. 6b). Previous reports have demonstrated that these new peaks correspond to the presence of 13C at the C2 position of D-(2-13C) mannose.24 This finding is in agreement with our HPLC results which demonstrated that mannose was the primary product of this reaction. Since no peaks at δ = 94.6 and 95.1 ppm corresponding to D-(1-13C) mannose were observed, our NMR results suggest that the hybrid catalyst converts D-(1-13C) glucose to D-(2-13C) mannose, demonstrating the catalyzed epimerization proceeded by a 1,2-carbon shift mechanism.
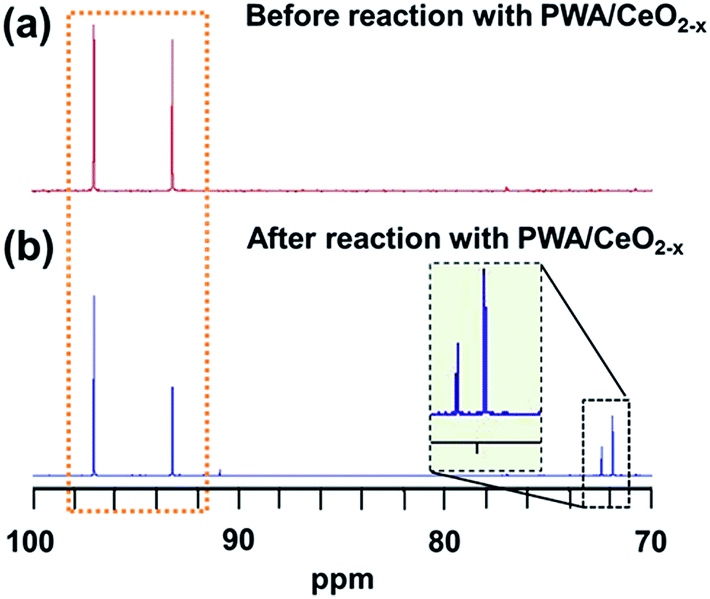 | ||
| Fig. 6 13C NMR spectra of D-(1-13C) glucose solution (a) before the reaction and (b) after the reaction. peaks corresponding to D-(1-13C) glucose are highlighted by an orange dotted rectangle. Peaks corresponding to D-(2-13C) mannose are highlighted by a black dashed rectangle. | ||
3.6 Reaction mechanism for mannose production
The results of the above catalytic activity studies lead us to hypothesize a two-step mechanism for the production of mannose from cellobiose catalyzed by the hybrid PWA/CeO2−x catalyst (Fig. 7). In the first step, cellobiose is hydrolyzed into glucose molecules by the supported PWA material. In the second step, the resulting glucose molecules are then epimerized to mannose via a 1,2-carbon shift catalyzed by cerium ions, which may have been leached from the cerium oxide support. This epimerization process allows for promoting the cellobiose conversion by converting a portion of the glucose products to mannose.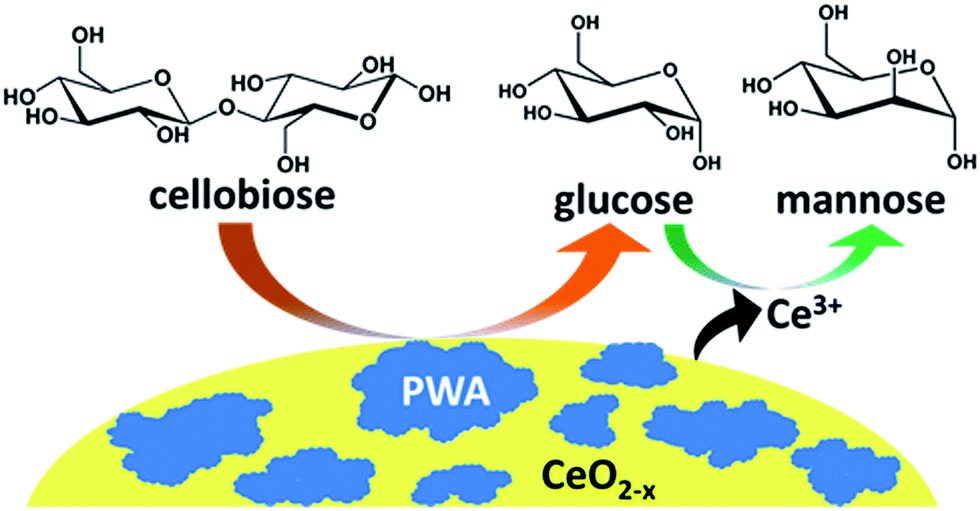 | ||
| Fig. 7 Two step conversion of cellobiose to mannose by the PWA/CeO2−x catalyst. In the first step, cellobiose is hydrolyzed to form glucose monomers. In the second step, the glucose monomers are converted to mannose by the leached cerium ions. | ||
4. Conclusions
A hybrid PWA/CeO2−x catalyst material was demonstrated to effectively convert cellobiose to mannose and epimerize glucose to mannose. The catalytic activity of the material for cellobiose hydrolysis was illustrated over a range of reaction temperatures and durations to primarily yield glucose and mannose. When the catalytic activity of each catalyst component was discretely investigated, the combined hybrid catalyst was inferred to display a synergistic effect for the conversion of cellobiose. Our findings suggest that this effect indirectly stemmed from the epimerization of glucose to mannose. Using 13C NMR, we further demonstrated that this epimerization proceeded by the way of a 1,2-carbon shift mechanism similar to that previously reported for several metal ion catalyst systems.12 Upon closer investigation, the hybrid catalyst's primary method for causing this epimerization was found to be likely through homogeneous catalysis of leached cerium ions. The ability of the hybrid catalyst to cause epimerization under acidic conditions allows it to avoid the formation of 2-ketoses which are typically formed under basic conditions. The understanding gained in this study is expected to promote the development of improved catalysts for rare sugar production from cellulose. Although this catalyst has been demonstrated to be effective for cleaving β-(1,4)-glycosidic bonds, its application towards cellulose will likely require the use of other solvent systems which can more readily solubilize cellulose crystals. We propose the investigation of ionic liquids for this purpose due to their successful application to many other solid acid catalyst systems.40,41 Finally, our study demonstrates that as more supported PWA catalysts are developed, the presence and effect of support leaching on catalytic activity should be closely investigated.Acknowledgements
The authors acknowledge funding support from the Nebraska Center for Energy Sciences and Army Research Office (W911NF-10-2-0099). JJB and KOS acknowledge the support of the UCARE program at the University of Nebraska-Lincoln (UNL). JJB is also grateful for the NASA Nebraska Space Grant Fellowship support. We thank the Undergraduate Instrumentation Center and Research Instrumentation Facility in the Department of Chemistry, Nebraska Center for Materials and Nanoscience, and the Morrison Microscopy Core Research Facility at UNL for the use of their facilities. The authors also acknowledge Michael Tai and Deepak Keshwani for help with HPLC verification of mannose in our samples and Martha Morton for help with NMR analysis. We thank Joseph Brewer and Josh Beaudoin for help with ICP-MS measurements. In addition, we thank Johnny Goodwin at the University of Alabama Central Analytical Facility for the high resolution electron microscopy work.Notes and references
- M. S. Singhvi, S. Chaudhari and D. V. Gokhale, RSC Adv., 2014, 4, 8271–8277 RSC.
- L. Sunggyu and W. Barbara, in Handbook of Alternative Fuel Technologies, CRC Press, 2nd edn, 2014, pp. 1–18 Search PubMed.
- M. Balat and H. Balat, Appl. Energy, 2009, 86, 2273–2282 CrossRef CAS.
- Y. H. P. Zhang, J. B. Cui, L. R. Lynd and L. R. Kuang, Biomacromolecules, 2006, 7, 644–648 CrossRef CAS PubMed.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- P. Alvira, E. Tomas-Pejo, M. Ballesteros and M. J. Negro, Bioresour. Technol., 2010, 101, 4851–4861 CrossRef CAS PubMed.
- C. Chatterjee, F. Pong and A. Sen, Green Chem., 2015, 17, 40–71 RSC.
- S. Angyal, in Glycoscience, ed. A. Stütz, Springer, Berlin Heidelberg, 2001, vol. 215, pp. 1–14 Search PubMed.
- C.-S. Park, H.-J. Kwon, S.-J. Yeom and D.-K. Oh, Biotechnol. Lett., 2010, 32, 1305–1309 CrossRef CAS PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- S. Nguyen, S. Sophonputtanaphoca, E. Kim and M. Penner, Appl. Biochem. Biotechnol., 2009, 158, 352–361 CrossRef CAS PubMed.
- L. Petruš, M. Petrušová and Z. Hricovíniová, in Glycoscience, ed. A. Stütz, Springer, Berlin, Heidelberg, 2001, vol. 215, pp. 15–41 Search PubMed.
- Y. B. Huang and Y. Fu, Green Chem., 2013, 15, 1095–1111 RSC.
- R. Rinaldi and F. Schuth, Energy Environ. Sci., 2009, 2, 610–626 CAS.
- Y. Zheng, Z. Pan and R. Zhang, Int. J. Agric. Biol. Eng., 2009, 2, 51–68 CAS.
- Z. H. Zhang and Z. B. K. Zhao, Carbohydr. Res., 2009, 344, 2069–2072 CrossRef CAS PubMed.
- A. Wang, C. Li, M. Zheng and T. Zhang, in The Role of Green Chemistry in Biomass Processing and Conversion, John Wiley & Sons, Inc., 2012, pp. 313–348 Search PubMed.
- W. P. Deng, Q. H. Zhang and Y. Wang, Dalton Trans., 2012, 41, 9817–9831 RSC.
- J. Tian, J. H. Wang, S. Zhao, C. Y. Jiang, X. Zhang and X. H. Wang, Cellulose, 2010, 17, 587–594 CrossRef CAS.
- M. Zhang, W. S. Zhu, S. H. Xun, H. M. Li, Q. Q. Gu, Z. Zhao and Q. Wang, Chem. Eng. J., 2013, 220, 328–336 CrossRef CAS.
- M. I. Alam, S. De, S. Dutta and B. Saha, RSC Adv., 2012, 2, 6890–6896 RSC.
- Á. Kukovecz, Z. Balogi, Z. Kónya, M. Toba, P. Lentz, S. I. Niwa, F. Mizukami, Á. Molnár, J. B. Nagy and I. Kiricsi, Appl. Catal., A, 2002, 228, 83–94 CrossRef.
- T. Tanase, T. Takei, M. Hidai and S. Yano, Carbohydr. Res., 2001, 333, 303–312 CrossRef CAS PubMed.
- W. R. Gunther, Y. Wang, Y. Ji, V. K. Michaelis, S. T. Hunt, R. G. Griffin and Y. Román-Leshkov, Nat. Commun., 2012, 3, 1109 CrossRef PubMed.
- R. Bermejo-Deval, R. Gounder and M. E. Davis, ACS Catal., 2012, 2, 2705–2713 CrossRef CAS.
- V. Choudhary, A. B. Pinar, R. F. Lobo, D. G. Vlachos and S. I. Sandler, ChemSusChem, 2013, 6, 2369–2376 CrossRef CAS PubMed.
- C. Sun, H. Li and L. Chen, Energy Environ. Sci., 2012, 5, 8475–8505 CAS.
- L. Vanoye, M. Fanselow, J. D. Holbrey, M. P. Atkins and K. R. Seddon, Green Chem., 2009, 11, 390–396 RSC.
- L. Negahdar, J. U. Oltmanns, S. Palkovits and R. Palkovits, Appl. Catal., B, 2014, 147, 677–683 CrossRef CAS.
- K. Li, L. Bai, P. N. Amaniampong, X. Jia, J.-M. Lee and Y. Yang, ChemSusChem, 2014, 7, 2670–2677 CrossRef CAS PubMed.
- X. Peng, X.-G. Meng, C. Mi and X.-H. Liao, RSC Adv., 2015, 5, 9348–9353 RSC.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter and D. Templeton, Determination of Sugars, Byproducts, and Degradation Products in Liquid Fraction Process Samples, U.S. Department of Energy, Golden, 2006 Search PubMed.
- V. P. Ananikov, K. A. Gayduk, I. P. Beletskaya, V. N. Khrustalev and M. Y. Antipin, Eur. J. Inorg. Chem., 2009, 2009, 1149–1161 CrossRef.
- E. Caliman, J. A. Dias, S. C. L. Dias, F. A. C. Garcia, J. L. de Macedo and L. S. Almeida, Microporous Mesoporous Mater., 2010, 132, 103–111 CrossRef CAS.
- J. Pang, A. Wang, M. Zheng, Y. Zhang, Y. Huang, X. Chen and T. Zhang, Green Chem., 2012, 14, 614–617 RSC.
- R. Rinaldi and F. Schuth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS PubMed.
- S. S. Pröckl, W. Kleist, M. A. Gruber and K. Köhler, Angew. Chem., Int. Ed., 2004, 43, 1881–1882 CrossRef PubMed.
- N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS.
- A. Ignatyev Igor, V. Doorslaer Charlie, G. N. Mertens Pascal, K. Binnemans and E. d. Vos Dirk, in Holzforschung, 2012, vol. 66, pp. 417–425 Search PubMed.
- Z. Zhang and Z. K. Zhao, Carbohydr. Res., 2009, 344, 2069–2072 CrossRef CAS PubMed.
- S.-J. Kim, A. A. Dwiatmoko, J. W. Choi, Y.-W. Suh, D. J. Suh and M. Oh, Bioresour. Technol., 2010, 101, 8273–8279 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A table comparing the % conversion and yields of product from reactions carried out at 160 °C for 60, 120, and 360 minutes in Teflon-lined stainless steel autoclaves heated by convection oven. See DOI: 10.1039/c5ra02645h |
| This journal is © The Royal Society of Chemistry 2015 |