
Mechanism study of Li+ insertion into titania nanotubes†
Abstract
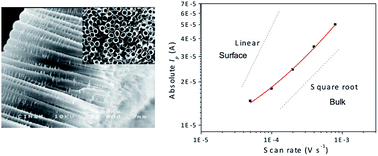
Li+ insertion into anatase titania nanotubes (TiO2nts) employing PEO-based polymer electrolyte has been studied by cyclic voltammetry and chronoamperometry. The study shows that Li+ storage in the anatase is dominated by the bulk diffusion (into the lattice) and the increasing contribution of the pseudo-capacitive effect with faster kinetics. We also report that the chemical diffusion of Li+ in self-organized TiO2nts is around 2 × 10−16 cm2 s−1 suggesting that the use of a solid electrolyte does not alter the charge transport in the nanostructured electrode.
 Please wait while we load your content...
Please wait while we load your content...

