
The role of cerium(iii)/yttrium(iii) nitrate hexahydrate salts on electroactive β phase nucleation and dielectric properties of poly(vinylidene fluoride) thin films
Abstract
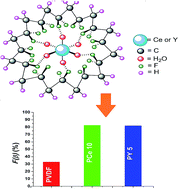
Electroactive poly(vinylidene fluoride) (PVDF) thin films modified with Ce(NO3)3·6H2O and Y(NO3)3·6H2O (1–30 mass%) have been prepared via a simple solution casting method. The thermal stability and microstructures of the films were investigated using thermal gravimetric analysis techniques and field emission electron microscopy respectively. X-ray diffraction spectroscopy, Fourier transform infrared spectroscopy and differential scanning calorimetry confirmed the nucleation of the electroactive β phase in the composite films. Strong interfacial interaction i.e. ion dipole interaction via formation of hydrogen bonds between the water molecules of the salts and the polar –CF2 dipoles of the polymer chains resulted in improved electroactive β phase nucleation and a large dielectric constant in PVDF films.
 Please wait while we load your content...
Please wait while we load your content...

