
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Utility of a heterogeneous palladium catalyst for the synthesis of a molecular semiconductor via Stille, Suzuki, and direct heteroarylation cross-coupling reactions†
Seth M.
McAfee
,
Jenny S. J.
McCahill
,
Casper M.
Macaulay
,
Arthur D.
Hendsbee
and
Gregory C.
Welch
*
Department of Chemistry, Dalhousie University, 6274 Coburg Road, P.O. Box 15000, Halifax, Nova Scotia, Canada B3H 4R2. E-mail: gregory.welch@dal.ca; Fax: +1 902 494 1310; Tel: +1 902 494 4245
First published on 27th February 2015
Abstract
The commercially available silica-supported heterogeneous catalyst SiliaCat® DPP-Pd has proven to be highly active, robust, and reusable for the synthesis of a thiophene–phthalimide-based molecular semiconductor under microwave-irradiation reaction conditions. A Stille reaction protocol demonstrated that SiliaCat® DPP-Pd outperformed well-known homogeneous catalysts, Pd(PPh3)4 and Pd(PPh3)2Cl2, in terms of performance and catalyst loading, while also exhibiting tolerance to ambient reaction conditions and two-fold recyclability for the formation of product. The success established for SiliaCat® DPP-Pd catalyzed Stille reactions via microwave irradiation was extended to optimize Suzuki coupling and direct heteroarylation protocols. Notably, direct heteroarylation with SiliaCat® DPP-Pd exhibited excellent selectivity and perturbed the formation of homo-coupled aryl bromides, two side reactions that are known to plague this type of cross-coupling reaction.
1. Introduction
Organic electronics is an ever-expanding field of study that makes use of organic materials as active components in electrical devices.1 Such devices are promoted to be ultra-low cost, light weight and flexible.2,3 Key technologies include field effect transistors, sensors, photovoltaics, and light-emitting devices.4–6 While great improvements in device performance have been realized over the past few years,7,8 only recently has the concept of ‘green electronics’ gained significant attention.9,10 To this end, key to the growth of organic electronics is the development of cost-effective organic semiconductors that can be readily accessible from raw materials, through a sustainable synthesis with scalable purification and easily processable from solution at low temperatures.11–15These organic semiconductors consist of delocalized π-conjugated building blocks. Their synthesis almost universally involves carbon–carbon bond forming reactions that rely on late transition metal catalysts (Ni, Pd, Pt) specifically, palladium is most commonly employed.11,16,17 Homogeneous palladium catalysts, typically stabilized by phosphine ligands, are highly sensitive to oxygen, moisture and high temperatures. The instability of such catalysts restricts their utility to inert atmosphere conditions with rigorously deoxygenated solvents, and renders them to single use.18–20 Separation of these homogeneous metal catalysts from the organic product is challenging but is of utmost importance to prevent trace metal contamination, which leads to the degradation of device performance.21–24
With the organic electronics industry rapidly expanding, kilograms to metric tons of organic semiconductors will be required to meet the demand. With the current costs of Pd and the inert atmosphere conditions required, new catalytic methods are desired for large-scale materials production. Commercially available heterogeneous catalysts offer a promising alternative to address these issues, where a Pd metal or a Pd complex with ligands can be directly fixed to a solid support such as carbon or silica, and offers numerous advantages.18,25–30 They are known to be more tolerant to temperature, can be utilized in air, are easily isolated from the reaction mixture and can be recycled for multiple uses.18,20,31–33 Unfortunately, heterogeneous catalysts are often less active and selective than their homogenous counter parts, and so they have not been extensively explored. To date, there is only one report of an high performance organic semiconductor being synthesized by heterogeneous catalysts, a conjugated polymer, synthesized using Pd/carbon,19 but the potential remains for future development of this synthetic protocol.
To coincide with industrially relevant practices and a streamlined synthetic procedure, microwave activation has been pursued as a means to reduce reaction times and improve yields, owing to a more uniform heating of the reaction mixture.34,35 Microwave reactors have been successfully implemented for the synthesis of many π-conjugated materials;36–41 however, the potential for microwave-assisted synthesis with heterogeneous catalysis has been scantly explored,32,42 and remains largely underappreciated. The combination of solid catalysts and microwave activation has been described to have several beneficial features, leading to a rapid, effective, and environmentally benign synthesis.42 Motivation for this synthetic protocol is also attributed to an increased purity in the final material, with microwave irradiation diminishing the formation of undesired by-products and heterogeneous Pd catalyst vastly reducing the instances of metal contamination.32
Despite this noted success, there has not been any substantial follow-up to this intriguing possibility as an industrially relevant synthetic method. In an effort to emphasize the prospective widespread application of heterogeneous catalysts herein we describe the application of these catalysts in direct comparison to their homogeneous counterparts for the synthesis of a high performance organic semiconductor accessible from a straightforward synthetic procedure via microwave irradiation.
2. Results and discussion
The utility of a heterogeneous catalyst has been assessed for three prevalent palladium catalyzed cross-couplings: Stille reaction,17 Suzuki coupling,43,44 and direct heteroarylation.45 To fully explore the efficacy of a heterogeneous catalyst for the synthesis of a molecular semiconductor via Stille reaction we evaluated several key parameters: (1) performance in comparison to homogeneous alternatives, (2) catalyst loadings, (3) sensitivity to ambient reaction conditions, and (4) recyclability of the catalyst. Considering the promising results, we extended our optimized synthesis with heterogeneous catalysts to both Suzuki and direct heteroarylation protocols and assessed their performance.As alluded to earlier, the current procedure for the synthesis of molecular semiconductors has been trending from the conventional method of oil bath heating towards microwave-assisted synthesis, which has been shown to offer a rapid and high yielding synthesis. To ensure our results are as relevant as possible to this rapidly evolving field we first optimized microwave-heating conditions (see Experimental section) to be used for the majority of our synthetic procedure with oil bath heating used only for select reaction conditions.
Our choice of target molecule, 5,5′(2,2′-bithiophene-5,5′-diyl)bis(2-hexylphthalimide) (SM-1, Fig. 1) was influenced by recent publications from our group on phthalimide–thiophene-based molecular semiconductors.36,46 These materials yield high electron mobilities in thin-film transistors, are low-cost and can be easily accessed via a straightforward synthesis from low-cost and readily available building blocks, and thus represents an ideal molecular design for the purpose of our study.
 | ||
| Fig. 1 Structure of the target molecule, 5,5′(2,2′-bithiophene-5,5′-diyl)bis(2-hexylphthalimide). | ||
Furthermore, the ease of synthesis and straightforward isolation of SM-1 highlighted the potential to use this material for a catalyst study. The formation of the orange product from colourless starting materials allows for the straightforward identification of a successful reaction, the material can be isolated by filtration through silica-gel to remove most inorganic impurities. 1H NMR spectral analysis of the material product was used to assess the extent of starting material conversion to product. Remaining starting material and residual inorganic and organic impurities are often the cause of greater than quantitative yields of the product material. These impurities can be easily removed via flash column chromatography with CH2Cl2 as the eluent; however, this was not required to simply investigate the conversion of starting material to product.
2.1 Stille reaction
The synthesis of the target molecule, SM-1, from synthesized starting materials (1, 2a) using commercial heterogeneous and homogeneous catalysts under microwave irradiation was investigated via Stille reaction (Scheme 1).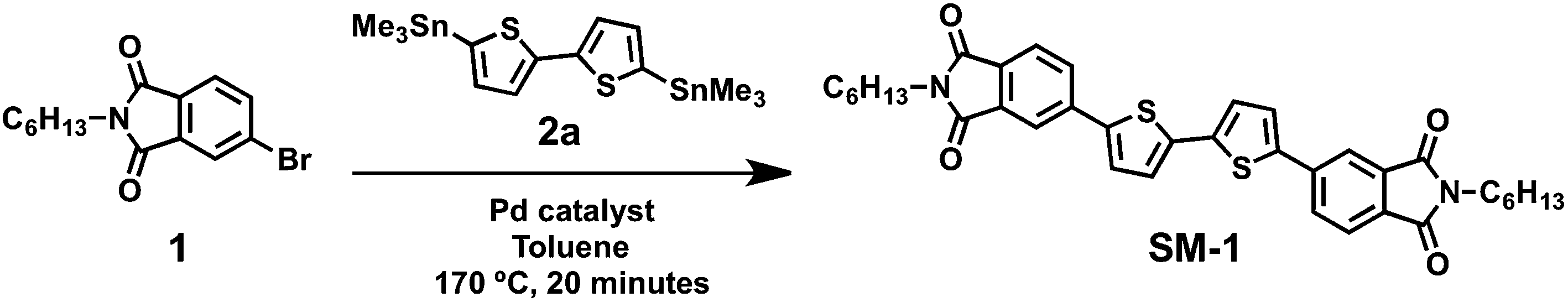 | ||
| Scheme 1 Reaction scheme for the synthesis of target molecule, SM-1, via palladium catalyzed cross coupling Stille reaction under optimized microwave irradiation conditions. | ||
Analysis of the 1H NMR spectra shows that all four catalysts were able to convert starting material to SM-1, albeit with varying degrees of success (Fig. 2). The material product obtained using Pd(PPh3)4 as a catalyst was shown to be high yielding, with SM-1 as the sole product based on 1H NMR spectroscopy, while the product obtained using Pd(PPh3)2Cl2 was realized in higher than quantitative yields, and can be attributed to baseline impurities and residual starting material. The SiliaCat® DPP-Pd catalyzed reaction returned product in high yield and the 1H NMR spectrum of the material is consistent with SM-1 as the major product with small amounts of baseline impurities. Lastly, SiliaCat® S-Pd as a catalyst for the Stille reaction resulted in an incomplete reaction, accompanied by a low yield and a product material with significant impurities and starting material present. Using the conditions outlined above Pd(PPh3)4, Pd(PPh3)2Cl2 and SiliaCat® DPP-Pd were found to be effective catalysts for the Stille reaction with isolation of the desired product in high yield. Based on the low yield and large product impurity obtained using the SiliaCat® S-Pd, the optimization of the Stille reaction was further investigated with just the one heterogeneous catalyst, SiliaCat® DPP-Pd.
 | ||
| Fig. 2 Aromatic and aliphatic regions of the 1H NMR spectra of material product with varying homogeneous (1 & 3) and heterogeneous (4 & 5) Pd-catalysts at 5.0 mol% catalyst loading, synthesized via microwave irradiation. Material yield obtained after work up described in Experimental section, percent yield calculated as mass obtained/mass expected. Top spectrum (1) shows analytically pure SM-1. | ||
 | ||
| Fig. 3 1H NMR spectra of resultant products with varying Pd catalyst loadings (2–5) of SiliaCat® DPP-Pd. Top spectrum (1) shows analytically pure SM-1. | ||
Decreasing the SiliaCat® DPP-Pd loading from 5.0 mol% to 0.1 mol% had little impact on the product formation. At all catalyst loadings, SM-1 is the major product with only small quantities of baseline impurities, while the greater than quantitative yields for 0.1 mol% catalyst loading is attributed to unreacted starting material impurities.
In further comparison to homogeneous alternatives, the use of 1.0 mol% Pd loadings for Pd(PPh3)4 and Pd(PPh3)Cl2 were also investigated (Fig. 4).
 | ||
| Fig. 4 1H NMR spectra of resultant products using heterogeneous (2) and homogeneous (3 & 4) Pd catalysts at 1.0 mol% Pd loading. Top spectrum (1) shows analytically pure SM-1. | ||
Using a 1.0 mol% Pd loading both SiliaCat® DPP-Pd and Pd(PPh3)4 return high yields and SM-1 as the major product; however, the use of 1.0 mol% Pd loading of Pd(PPh3)2Cl2 as a catalyst results in an incomplete reaction where the product material contained significant impurities and unreacted starting material, contributing to the greater than quantitative yield observed, and therefore was not used in subsequent experiments.
 | ||
| Fig. 5 1H NMR spectra of resultant products using 1.0 mol% SiliaCat® DPP-Pd loadings via microwave irradiation under inert (2) or ambient (3) conditions and conventional heating under ambient conditions (4). Top spectrum (1) shows analytically pure SM-1. | ||
At 1.0 mol% catalyst loadings, the microwave-assisted synthesis of SM-1 under inert or ambient reaction conditions return nearly identical 1H NMR spectra, accompanied by residual baseline impurities. The synthesis of SM-1 under ambient conditions via conventional heating methods did not show full conversion of the starting material, as indicated by 1H NMR spectroscopy. Considering this, the reaction was repeated at 5.0 mol% catalyst loading and 1H NMR confirmed a more complete reaction (Fig. S5†), and therefore we attribute the poor conversion at 1.0 mol% to slow catalyst degradation over the period of 24 hours under ambient conditions.
As described beforehand, heterogeneous catalysts are known to leave minimal trace Pd metal impurities in the products of their reactions. To evaluate the Pd leeching at 5.0 mol% catalyst loading under the conventional heating protocol, the product material was analyzed by ICP-OES. The SiliaCat® DPP-Pd catalyzed reaction supported this notion, the product material was found to contain 0.08 mg L−1 Pd in comparison to 26.25 mg L−1 Pd content for the corresponding Pd(PPh3)4 catalyzed reaction (Table S1†). Instrument and experimental details for this analysis can be found in the ESI.†
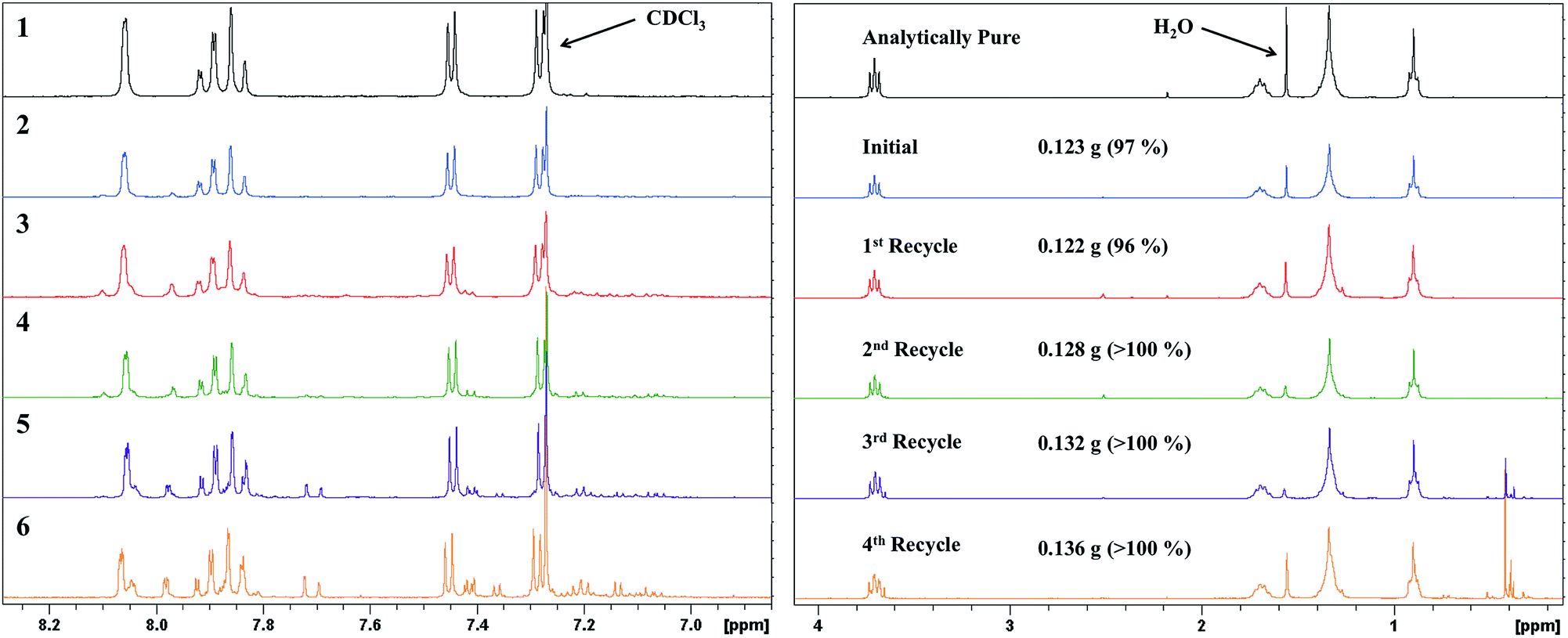 | ||
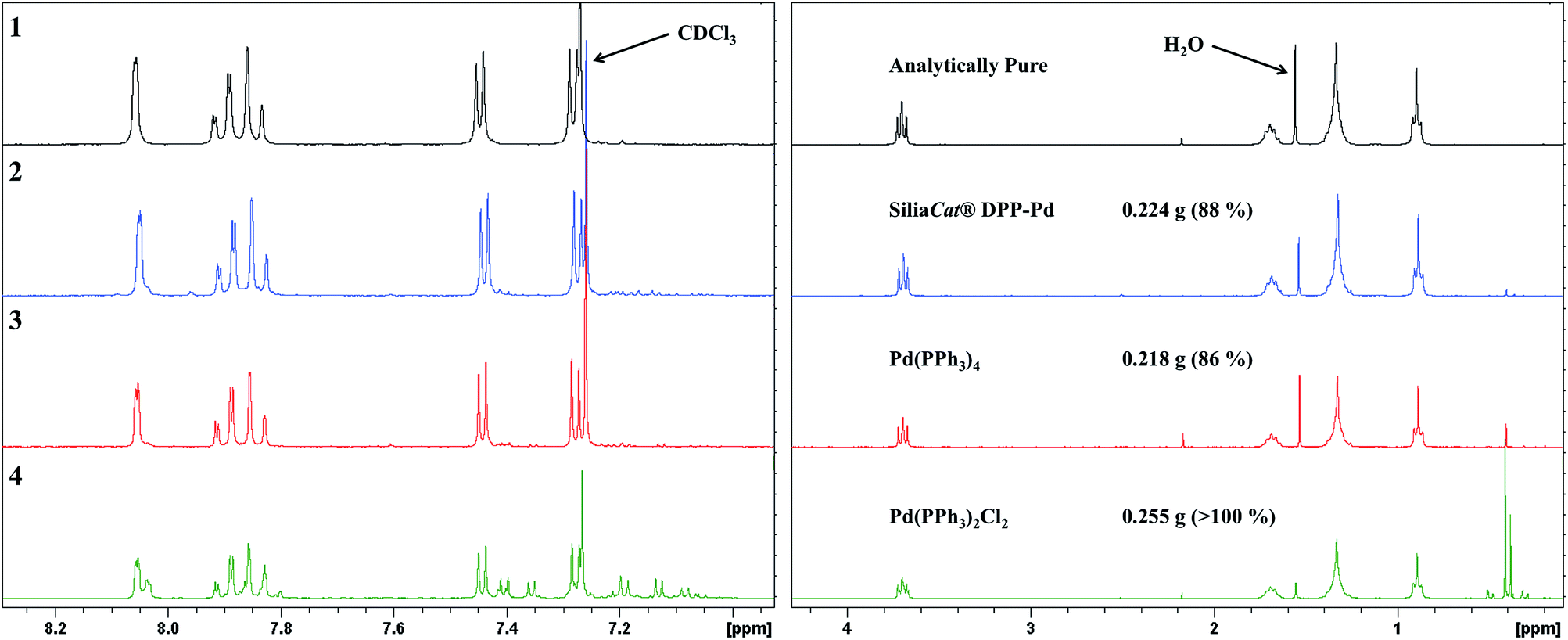
| Fig. 6 1H NMR spectra of resultant product from initial (2), and four-fold recyclability (3–6) trials of SiliaCat® DPP-Pd at 5.0 mol% catalyst loading, synthesized via microwave irradiation. Top spectrum (1) shows analytically pure SM-1. | ||
The recyclability of the catalyst was assessed for five consecutive reactions, with each trial after the initial using the recovered catalyst for the next reaction. We began at 5.0 mol% catalyst loadings and found that it was possible to recover roughly 90% of the catalyst from each trial. The 1H NMR spectra of the product material revealed that the catalyst can be effectively recycled twice to produce similar yields with residual starting material impurities contributing to the more than quantitative yield obtained after the second recycle. The third and fourth time the catalyst was recycled there remained a significant amount of starting material, contributing to the reported yields greater than 100%. These results suggest that the catalyst can be effectively recycled for re-use; however, beyond the second recycle there is a drop in conversion of starting material to product noted by the presence of significant baseline impurities.
2.2 Suzuki coupling
The synthesis of the target molecule, SM-1, from synthesized (1) and commercially available (2b) starting materials using the best performing heterogeneous catalyst, SiliaCat® DPP-Pd, under microwave irradiation with lab grade reagents was investigated via Suzuki cross-coupling reaction (Scheme 2). | ||
| Scheme 2 Reaction scheme for the synthesis of the target molecule, SM-1, via SiliaCat® DPP-Pd catalyzed Suzuki cross coupling reaction under optimized microwave irradiation conditions. | ||
 | ||
| Fig. 7 1H NMR spectra of resultant product for the optimization of Suzuki couplings, varying temperature (2–4), with 5.0 mol% catalyst loading of SiliaCat® DPP-Pd. Top spectrum (1) shows analytically pure SM-1. | ||
Unfortunately, full conversion of starting material was not observed for the Suzuki couplings under our selected conditions. We understand that the success of Suzuki couplings are often quite sensitive to the choice of solvent and base, perhaps further optimization of these conditions would yield improved conversion of starting material to product, this; however, was not the purpose of this study. Nevertheless, the product material can be quickly purified via flash column chromatography to give an analytically pure material while still maintaining an acceptable yield of final material (63%, in reference to moles of 2b).
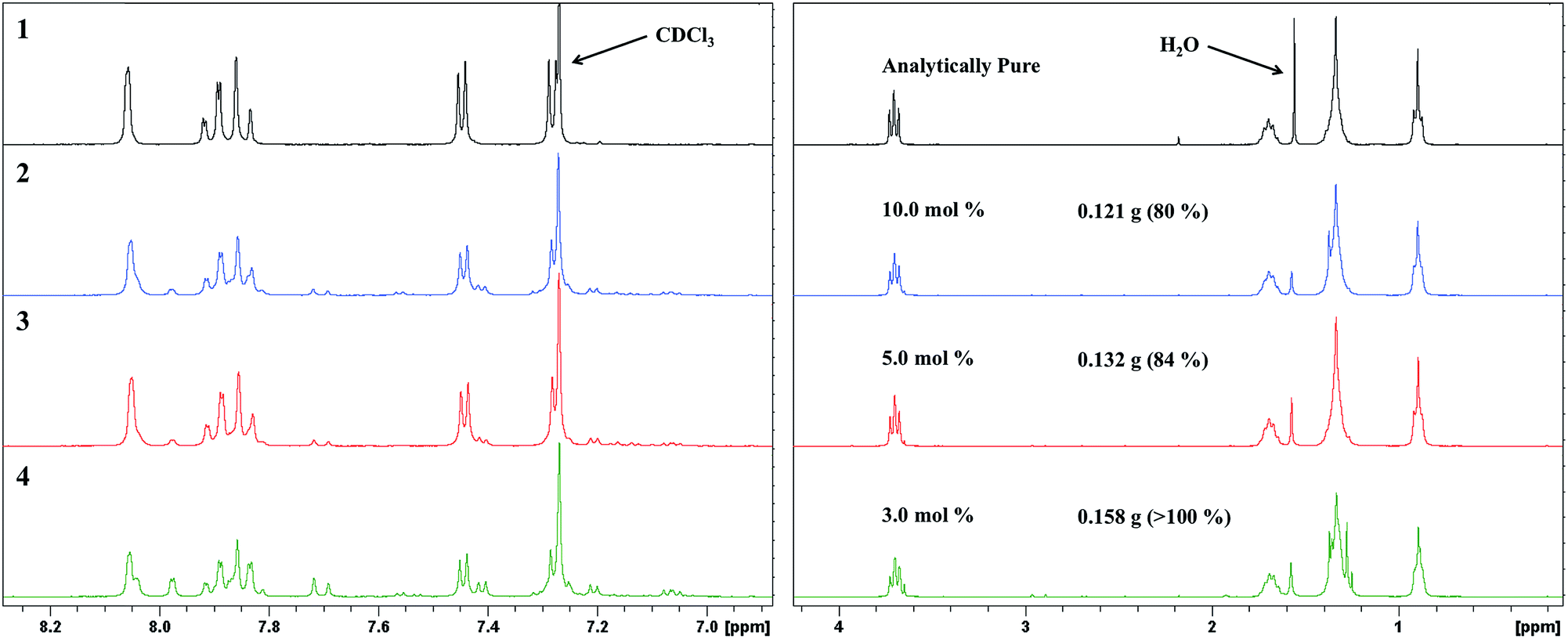 | ||
| Fig. 8 1H NMR spectra of resultant product with varying Pd loadings of SiliaCat® DPP-Pd (2–4), there was no product obtained from 1.0 mol% catalyst loading. Top spectrum (1) shows analytically pure SM-1. | ||
Decreasing the SiliaCat® DPP-Pd loading had a significant impact on the resultant product yield. The 1H NMR spectra for the reactions using 10.0 and 5.0 mol% loading (Fig. 8) show that SM-1 is the major product with only small quantities of starting material and baseline impurities. Decreasing the Pd loading to 3.0 mol% results in a product material with a larger amount of baseline impurity and starting material, contributing to the high yield, furthermore, 1.0 mol% did not result in any obtained final product. Considering these results, the Suzuki coupling reaction is highly dependent on catalyst loading, where loadings less than 5.0 mol% are not sufficiently active to convert starting material to product before the aforementioned decomposition of the boronate ester functionality. On the other hand, catalyst loadings greater than 5.0 mol% did not result in a significant increase in yield or conversion to final material.
2.3 Direct heteroarylation
The synthesis of the target molecule, SM-1, from synthesized starting materials (1 and 2c) using the common catalyst Pd(OAc)2 and the best performing heterogeneous catalyst, SiliaCat® DPP-Pd, under microwave irradiation with lab grade reagents was investigated via direct heteroarylation (Scheme 3).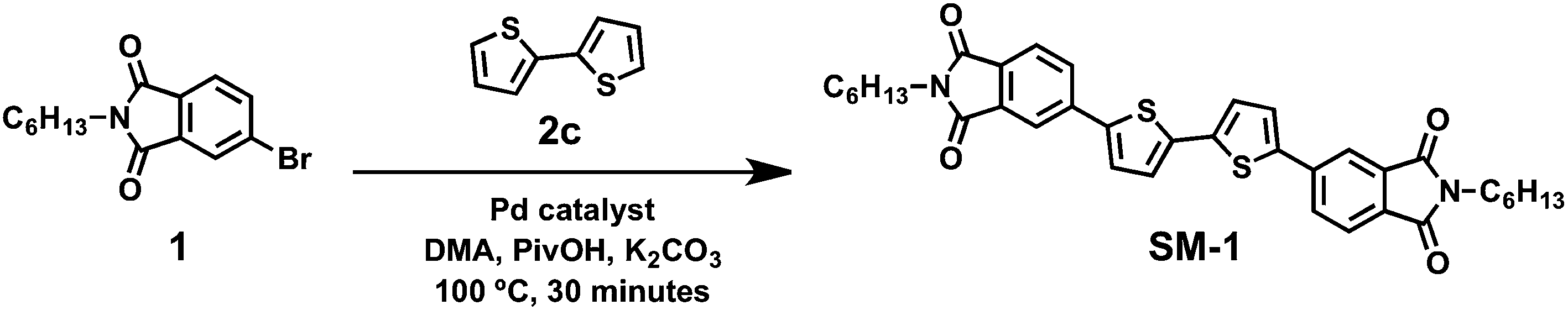 | ||
| Scheme 3 Reaction scheme for the synthesis of the target molecule, SM-1, via palladium catalyzed direct heteroarylation reaction under optimized microwave irradiation conditions. | ||
The advantages of direct heteroarylation over more traditional cross-coupling techniques have been well documented;52–55 with particular emphasis on ligand-free catalysts, no organotin or organoboron by-products and excellent atom economy. Despite these favourable characteristics, there remain some underlying issues that have impeded the widespread use of direct heteroarylation. The lack of directing groups induces complications pertaining to the selectivity of C–H bonds, leading to unwanted isomers, or cross-linked materials during polymerization reactions.45 While another side reaction that typically plagues phosphine-free direct heteroarylation is the homo-coupling of aryl bromides, which has been shown to be more prevalent at higher temperatures.56 It has been proposed that fine-tuning of reaction conditions, such as temperature, and reaction time may subdue these other products. Considering the successful use of supported Pd catalysts for intramolecular direct heteroarylation,57 we sought to investigate the potential use of SiliaCat® DPP-Pd for intermolecular direct heteroarylation with optimized reaction conditions to preclude the formation homo-coupled aryl bromides, while also achieving the desired selectivity.
| Catalyst | Time (min) | Material | Yield |
SM-1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3 :2:3 |
|---|---|---|---|---|
| Pd(OAc)2 | 10 | 0.134 g | 69% | 1:trace:0.3 |
| SiliaCat® DPP-Pd | 10 | 0.058 g | 30% | 1:2.3:trace |
| SiliaCat® DPP-Pd | 30 | 0.168 g | 87% | 1:0.3:trace |
 | ||
| Fig. 9 1H NMR aromatic spectra of resultant material obtained from SiliaCat® DPP-Pd (4–5) and Pd(OAc)2 (6) catalyzed direct heteroarylation at 5.0 mol% catalyst loading, synthesized via microwave irradiation. Spectra 1–3 shows analytically pure SM-1, SM-2, and SM-3 respectively. | ||
Analysis of the product material 1H NMR spectra highlights the advantages of SiliaCat® DPP-Pd in comparison to Pd(OAc)2. The product obtained from the Pd(OAc)2 catalyzed reaction shows the formation of the desired material accompanied by the unwanted homo-coupled aryl bromide compound, SM-3. Notably, this does not occur under the same conditions for the SiliaCat® DPP-Pd catalyzed reaction, where instead the low yielding incomplete reaction shows a large formation of mono-substituted product, SM-2, and not the disproportionation product. When the reaction time was increased to 30 minutes we saw full conversion of the starting material to the desired product with residual baseline impurities, indicating a step-wise reaction. This serves to highlight the viability of SiliaCat® DPP-Pd for direct heteroarylation, where the homo-coupled aryl bromide product is not favoured under our optimized microwave irradiation conditions. Furthermore, SiliaCat® DPP-Pd demonstrated appropriate C–H bond selectivity, which was exclusive to the 2-position of the starting material, 2c, as confirmed by 2-D 1H NMR (Fig. S6†).
3. Conclusion
We have investigated the utility of commercially available heterogeneous catalyst, SiliaCat® DPP-Pd, for the formation of a high performance molecular semiconductor via three common carbon–carbon bond forming reactions.Stille reaction protocols have highlighted the excellent performance of SiliaCat® DPP-Pd in comparison to homogeneous catalyst alternatives and demonstrated effective conversion of starting material to product with catalyst loadings down to 0.1 mol%. SiliaCat® DPP-Pd proved to be tolerant to both lab grade reagents and the capacity for two-fold recyclability without any significant impact on the material product and the respective yield.
Suzuki couplings with SiliaCat® DPP-Pd proved to be sluggish, where acceptable product formation was only achieved at moderate temperatures and with high catalyst loading required to obtain an acceptable conversion of starting material to the desired product.
SiliaCat® DPP-Pd was remarkably effective for direct heteroarylation-type reactions. The use of a heterogeneous catalyst in place of the customary homogeneous catalyst, Pd(OAc)2, significantly reduced the formation of homo-coupled aryl bromides while also demonstrating a more selective ‘step-wise’ reaction, through which a mono-substituted compound is formed first, and thus requiring prolonged reaction times to obtain the desired bis-substituted product.
The robust and efficient performance of SiliaCat® DPP-Pd coupled with its capacity to be used and stored under ambient conditions should be extremely useful for both routine and selected synthesis of organic semiconductors relevant to the field of organic electronics, in particular photovoltaics and thin-film transistors.
4. Experimental
4.1 General synthetic details
Preparations were carried out on a bench top or under an atmosphere of dry, O2-free N2via Schlenk line techniques and/or an Innovative Technology Inc. N2 atmosphere glove box. All microwave reactions were carried out using a Biotage® Initiator+ microwave reactor. The operational power range of the instrument is 0–400 W, using a 2.45 GHz magnetron. Pressurized air was used to cool each reaction after microwave heating.4.2 Materials
Heterogeneous catalysts SiliaCat® DPP-Pd and S-Pd were provided by SiliCycle. Homogeneous catalysts Pd(PPh3)4 and Pd(PPh2)Cl2 were purchased from Strem Chemicals Inc. All other materials were purchased from either Sigma-Aldrich or TCI America.4.3 Characterization
1H NMR spectroscopy was used to evaluate conversion to product. 1H NMR spectra were recorded on Bruker Avance 300 MHz or 500 MHz spectrometer at 300 K. Chemical shifts are referenced to SiMe4, and all experiments performed in CDCl3.4.4 Starting materials synthetic procedure
Starting materials 2a58 and 2c59 were synthesized according to their respective literature procedures.4.5 Microwave Stille reaction synthetic procedure
4-Bromo-N-hexyl phthalimide (0.252 g, 0.81 mmol), 5,5′-bis(trimethylstannyl)-2,2′-bithiophene (0.200 g, 0.41 mmol) and Pd catalyst were loaded into a 2–5 mL microwave vial with a stir bar. 3 mL of anhydrous toluene was added as the solvent and the reaction vial sealed under inert (N2) atmosphere. The reaction vial was placed in a microwave and heated for 5 min at 100 °C and then 20 min at 170 °C. After the vial had cooled to ambient temperature the contents were washed from the vial using 100 mL of CH2Cl2. To this solution 2 g of 10% K2CO3–90% silica-gel was added and stirred for 10 minutes. The solution was filtered through a silica plug eluting with 200 mL of a 10% NEt3–90% CH2Cl2 solution. Solvent was removed using a rotary evaporator and the resultant solid collected via filtration and washed with a 50% aqueous MeOH solution. The collected solid was dried in vacuo and weighed to give material yield.4.6 Conventional Stille reaction synthetic procedure
4-Bromo-N-hexyl phthalimide (0.252 g, 0.81 mmol), 5,5′-bis(trimethylstannyl)-2,2′-bithiophene (0.200 g, 0.41 mmol) and Pd catalyst were loaded into a round bottom flask with a stir bar. 10 mL of lab grade toluene was added to the flask and the flask fitted with a reflux condenser. The reaction was heated to 110 °C for 24 h. After the flask was cooled to room temperature 50 mL of CH2Cl2 and 2 g of 10% K2CO3–90% silica-gel was added and stirred for 10 minutes. The solution was filtered through a silica plug eluting with 200 mL of a 10% NEt3–90% CH2Cl2 solution. Solvent was removed using a rotary evaporator and the resultant solid collected via filtration and washed with 50% methanol, 50% water solution. The collected solid was dried in vacuo and weighed to give material yield.4.7 Catalyst recyclability studies
Synthetic set up and reaction conditions were same as described in the microwave synthetic procedure. After the reaction vial had cooled the solution was poured into 50 mL of CH2Cl2. The solution was passed through a frit to isolate the supported catalyst. The recovered catalyst was washed with 50 mL of CH2Cl2, collected and dried in vacuo. The recovered catalyst was weighed and stored until further use. The filtrate was worked up as described previously above.4.8 Microwave Suzuki coupling synthetic procedure
4-Bromo-N-hexyl phthalimide (0.148 g, 0.48 mmol), 2,2′-bithiophene-5,5′-diboronic acid bis(pinacol) ester (0.100 g, 0.24 mmol), K2CO3 (0.079 g, 0.57 mmol) and SiliaCat® DPP-Pd catalyst were loaded into a 2–5 mL microwave vial with a stir bar. 4 mL of bench top N,N-dimethylformamide (DMF) was added as the solvent and the reaction vial sealed under inert (N2) atmosphere. The reaction vial was placed in a microwave reactor and heated for 30 min at 90 °C. After the vial had cooled to ambient temperature the contents were washed from the vial using 100 mL of CH2Cl2. To this solution 1 g of silica-gel was added and stirred for 10 minutes. The solution was filtered through a silica plug eluting with 200 mL of a 10% NEt3–90% CH2Cl2 solution. Solvent was removed using a rotary evaporator and the resultant solid was collected via filtration and washed with a 50% aqueous MeOH solution. The collected solid was dried in vacuo and weighed to give material yield.4.9 Microwave direct heteroarylation synthetic procedure
4-Bromo-N-hexyl phthalimide (0.188 g, 0.61 mmol), 2,2′-bithiophene (0.052 g, 0.31 mmol), potassium carbonate (0.208 g, 1.50 mmol), pivalic acid (0.023 g, 0.23 mmol) and SiliaCat® DPP-Pd catalyst were loaded into a 2–5 mL microwave vial with a stir bar. 4 mL of bench top N,N-dimethylacetamide (DMA) was added as the solvent and the reaction vial sealed under inert (N2) atmosphere. The reaction vial was placed in a microwave and heated for 30 min at 100 °C. After the vial had cooled to ambient temperature the contents were precipitated into 100 mL of H2O and allowed to stir for 1 hour. The solid precipitate was isolated by filtration and dissolved with 100 mL of CH2Cl2. To the filtrate, 1 g of silica-gel was added and stirred for 10 minutes. The solution was filtered through a silica plug eluting with 200 mL of a 10% NEt3–90% CH2Cl2 solution. Solvent was removed using a rotary evaporator and the resultant solid collected via filtration and washed with a 50% aqueous MeOH solution. The collected solid was dried in vacuo and weighed to give material yield.4.10 Analytical purification
The product material (0.207 g, 0.33 mmol) was dissolved in CH2Cl2 (50 mL) and liquid loaded onto a commercially available SiliaSep™ 25 g cartridge. Automated column chromatography was completed using a Biotage® Isolera flash system and a pentanes–CH2Cl2 gradient with the product eluting in 100% CH2Cl2. The product was isolated as an orange solid in 64% yield (0.130 g, 0.21 mmol) in reference to the initial material product.Acknowledgements
Dalhousie University, the Canada Research Chairs Program, and the Natural Science and Engineering Council of Canada (NSERC EGP 453320-2013) are acknowledged for financial support. Silicycle is acknowledged for supplying the catalysts for this study.References
- M. Berggren, D. Nilsson and N. D. Robinson, Nat. Mater., 2007, 6, 3 CrossRef CAS PubMed.
- S. R. Forrest, Nature, 2004, 428, 911 CrossRef CAS PubMed.
- M. Kaltenbrunner, T. Sekitani, J. Reeder, T. Yokota, K. Kuribara, T. Tokuhara, M. Drack, R. Schwödiauer, I. Graz, S. Bauer-Gogonea, S. Bauer and T. Someya, Nature, 2013, 499, 458 CrossRef CAS PubMed.
- T. W. Kelley, P. F. Baude, C. Gerlach, D. E. Ender, D. Muyres, M. A. Haase, D. E. Vogel and S. D. Theiss, Chem. Mater., 2004, 16, 4413 CrossRef CAS.
- A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3 CrossRef CAS PubMed.
- Y.-W. Su, S.-C. Lan and K.-H. Wei, Mater. Today, 2012, 15, 554 CrossRef CAS.
- I. McCulloch, Adv. Mater., 2013, 25, 1811 CrossRef CAS PubMed.
- J. E. Anthony, Nat. Mater., 2014, 13, 773 CrossRef CAS PubMed.
- M. Irimia-Vladu, E. D. Głowacki, G. Voss, S. Bauer and N. S. Sariciftci, Mater. Today, 2012, 15, 340 CrossRef CAS.
- M. Irimia-Vladu, Chem. Soc. Rev., 2013, 43, 588 RSC.
- G. Marzano, C. V. Ciasca, F. Babudri, G. Bianchi, A. Pellegrino, R. Po and G. M. Farinola, Eur. J. Org. Chem., 2014, 6583 CrossRef CAS PubMed.
- S. B. Darling and F. You, RSC Adv., 2013, 3, 17633 RSC.
- D. J. Burke and D. J. Lipomi, Energy Environ. Sci., 2013, 6, 2053 CAS.
- T. P. Osedach, T. L. Andrew and V. Bulović, Energy Environ. Sci., 2013, 6, 711 CAS.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31 RSC.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef CAS PubMed.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493 CrossRef CAS PubMed.
- L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133 CrossRef CAS PubMed.
- S.-Y. Liu, H.-Y. Li, M.-M. Shi, H. Jiang, X.-L. Hu, W.-Q. Li, L. Fu and H.-Z. Chen, Macromolecules, 2012, 45, 9004 CrossRef CAS.
- M. Pagliaro, V. Pandarus, R. Ciriminna, F. Béland and P. Demma Carà, ChemCatChem, 2012, 4, 432 CrossRef CAS PubMed.
- F. C. Krebs, R. B. Nyberg and M. Jørgensen, Chem. Mater., 2004, 16, 1313 CrossRef CAS.
- K. T. Nielsen, K. Bechgaard and F. C. Krebs, Macromolecules, 2005, 38, 658 CrossRef CAS.
- F. E. Alemdaroglu, S. C. Alexander, D. Ji, D. K. Prusty, M. Börsch and A. Herrmann, Macromolecules, 2009, 42, 6529 CrossRef CAS.
- Y. Jiang, T. Okamoto, H. A. Becerril, S. Hong, M. L. Tang, A. C. Mayer, J. E. Parmer, M. D. McGehee and Z. Bao, Macromolecules, 2010, 43, 6361 CrossRef CAS.
- M. Seki, Synthesis, 2006, 2006, 2975 CrossRef.
- A. Biffis, M. Zecca and M. Basato, J. Mol. Catal. A: Chem., 2001, 173, 249 CrossRef CAS.
- H.-U. Blaser, A. Indolese, A. Schnyder, H. Steiner and M. Studer, J. Mol. Catal. A: Chem., 2001, 173, 3 CrossRef CAS.
- V. Polshettiwar, C. Len and A. Fihri, Coord. Chem. Rev., 2009, 253, 2599 CrossRef CAS PubMed.
- G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Mülhaupt, J. Am. Chem. Soc., 2009, 131, 8262 CrossRef CAS PubMed.
- X. Chen, Y. Hou, H. Wang, Y. Cao and J. He, J. Phys. Chem. C, 2008, 112, 8172 CAS.
- Á. Molnár, Chem. Rev., 2011, 111, 2251 CrossRef PubMed.
- S. Alesi, F. D. Maria, M. Melucci, D. J. Macquarrie, R. Luque and G. Barbarella, Green Chem., 2008, 10, 517 RSC.
- F.-X. Felpin, T. Ayad and S. Mitra, Eur. J. Org. Chem., 2006, 2679 CrossRef CAS PubMed.
- C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250 CrossRef CAS PubMed.
- P. Lidström, J. Tierney, B. Wathey and J. Westman, Tetrahedron, 2001, 57, 9225 CrossRef.
- A. D. Hendsbee, C. M. Macaulay and G. C. Welch, Dyes Pigm., 2014, 102, 204 CrossRef CAS PubMed.
- R. C. Coffin, J. Peet, J. Rogers and G. C. Bazan, Nat. Chem., 2009, 1, 657 CrossRef CAS PubMed.
- S. J. Choi, J. Kuwabara and T. Kanbara, ACS Sustainable Chem. Eng., 2013, 1, 878 CrossRef CAS.
- M. Melucci, G. Barbarella and G. Sotgiu, J. Org. Chem., 2002, 67, 8877 CrossRef CAS PubMed.
- M. Melucci, G. Barbarella, M. Zambianchi, P. Di Pietro and A. Bongini, J. Org. Chem., 2004, 69, 4821 CrossRef CAS PubMed.
- W. Zhang, P. Lu, Z. Wang and Y. Ma, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1950 CrossRef CAS PubMed.
- A. Daştan, A. Kulkarni and B. Török, Green Chem., 2012, 14, 17 RSC.
- H.-H. Zhang, C.-H. Xing and Q.-S. Hu, J. Am. Chem. Soc., 2012, 134, 13156 CrossRef CAS PubMed.
- T. Yokozawa, H. Kohno, Y. Ohta and A. Yokoyama, Macromolecules, 2010, 43, 7095 CrossRef CAS.
- L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013, 46, 1597 CrossRef CAS PubMed.
- J.-P. Sun, A. D. Hendsbee, A. F. Eftaiha, C. Macaulay, L. R. Rutledge, G. C. Welch and I. G. Hill, J. Mater. Chem. C, 2014, 2, 2612 RSC.
- M. Lemay, V. Pandarus, M. Simard, O. Marion, L. Tremblay and F. Béland, Top. Catal., 2010, 53, 1059 CrossRef CAS.
- V. Pandarus, D. Desplantier-Giscard, G. Gingras, F. Béland, R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2013, 17, 1492 CrossRef CAS.
- V. Pandarus, G. Gingras, F. Béland, R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2012, 16, 117 CrossRef CAS.
- S. M. McAfee, J. M. Topple, A.-J. Payne, J.-P. Sun, I. G. Hill and G. C. Welch, ChemPhysChem, 2014 DOI:10.1002/cphc.201402662.
- G. A. Molander and N. Ellis, Acc. Chem. Res., 2007, 40, 275 CrossRef CAS PubMed.
- P. Berrouard, A. Najari, A. Pron, D. Gendron, P.-O. Morin, J.-R. Pouliot, J. Veilleux and M. Leclerc, Angew. Chem., Int. Ed., 2012, 51, 2068 CrossRef CAS PubMed.
- J. Kudrjasova, R. Herckens, H. Penxten, P. Adriaensens, L. Lutsen, D. Vanderzande and W. Maes, Org. Biomol. Chem., 2014, 12, 4663 CAS.
- J. Zhang, W. Chen, A. J. Rojas, E. V. Jucov, T. V. Timofeeva, T. C. Parker, S. Barlow and S. R. Marder, J. Am. Chem. Soc., 2013, 135, 16376 CrossRef CAS PubMed.
- D. J. Schipper and K. Fagnou, Chem. Mater., 2011, 23, 1594 CrossRef CAS.
- F. Lombeck, H. Komber, S. I. Gorelsky and M. Sommer, ACS Macro Lett., 2014, 3, 819 CrossRef CAS.
- M. Parisien, D. Valette and K. Fagnou, J. Org. Chem., 2005, 70, 7578 CrossRef CAS PubMed.
- C.-H. Cho, H. Kang, T. E. Kang, H.-H. Cho, S. C. Yoon, M.-K. Jeon and B. J. Kim, Chem. Commun., 2011, 47, 3577 RSC.
- C.-Y. Kuo, Y.-C. Huang, C.-Y. Hsiow, Y.-W. Yang, C.-I. Huang, S.-P. Rwei, H.-L. Wang and L. Wang, Macromolecules, 2013, 46, 5985 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complete NMR spectra presented. See DOI: 10.1039/c5ra02468d |
| This journal is © The Royal Society of Chemistry 2015 |