
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResolution of enantiomers with both achiral phases in chromatography: conceptual challenge
Ravi
Bhushan
*a,
Hariom
Nagar
a and
Jürgen
Martens
b
aDepartment of Chemistry, Indian Institute of Technology Roorkee, Roorkee – 247667, India. E-mail: rbushfcy54@gmail.com; hariomnagar@ymail.com; Fax: +91-1332-286202; Tel: +91-1332-285795
bInstitute of Chemistry, Carl von Ossietzky University Oldenburg, Carl-von-Ossietzky-Strasse 9-11, DE-26129 Oldenburg (Oldg), Germany. E-mail: juergen.martens@uni-oldenburg.de
First published on 12th March 2015
Abstract
A new practical approach has been adopted for the resolution (from racemic and non-racemic mixtures) of DL-selenomethionine using achiral phase chromatography supplemented with (−)-quinine as a chiral inducing reagent (CIR) and for the resolution of three racemic β-blockers (orciprenaline, betaxolol and atenolol) using L-Glutamic acid as a CIR. These analytes were pre-mixed with CIR and were applied to plain silica gel plates. The chromatograms were developed with the mobile phase having no chiral additive. Investigations were carried out by varying the pH, temperature and ratio of CIR to analytes. Native enantiomers were isolated and the detection limit for each enantiomer of SeMet was found to be 0.18 μg mL−1 and for enantiomers of the β-blockers it was found to be in the range of 1.2–1.8 μg mL−1. Optimized structures of transient diastereomers based on density functional theory using the Gaussian 09 Rev. A.02 program and hybrid density functional B3LYP with 6-31G* basis set have been developed. The separation mechanism has also been explained.
1. Introduction
As per IUPAC,1 the term ‘resolution’ in stereochemistry stands for a process, for the separation of racemic compounds (equimolar mixture of a pair of enantiomers) into their enantiomers. Formation of diastereomers is relevant for resolution either by chromatography or by (fractional) crystallization of diastereomeric salts. The term ‘resolution’ has always been used (in its classical sense in stereochemistry) for separation of a racemic mixture into its enantiomers, irrespective of the approach/methodology adopted. It may be more appropriate to use the term ‘separation’ if one is dealing with a non-racemic mixture.Resolution in an achiral environment or with both achiral phases in chromatography is not normally brought into consideration with the prevalent concepts of basic stereochemistry. In general, for a direct approach to resolution, the achiral phase in chromatography is brought into a chiral environment either by adding a suitable chiral selector to the mobile phase (CMPA) or by impregnating the stationary phase (the sorbent) with a chiral selector (e.g., in TLC).
Literature shows certain methods for separation of enantiomers from the excess enantiomer (i.e., non racemic mixture) under totally achiral conditions of physicochemical phase transitions or achiral chromatography. The sporadic reports till 1992 on such separations by liquid chromatography with both achiral phases were presented as a short review by Martens and Bhushan.2 Since then there appeared a few more reports on enantioresolution of certain non racemic mixtures in achiral environment, these included some antihistamines,3 binaphthol,4,5 certain heterocyclic compounds6 mandelic acid,7,8 different fluoro derivatives9–11 and amine derivatives.12
Martens and Bhushan13 addressed the basic question and the concept ‘why separation of one particular enantiomer (i.e., enantiomeric enrichment) should take place under achiral phase chromatography from the non-racemic mixture’, discussed briefly the technical terms used in literature and application of scientific terminology in the bounds of IUPAC while focusing on overlooked errors and scientific basis of separation in achiral environment during purification of enantiomeric mixtures in enantioselective synthesis.
The explanation, for separation under achiral conditions, considers preferential formation of homo- or hetero-associates (which could be dimeric, oligomeric, or polymeric) and one of them (e.g., the hetero-associate) is a high-order species having a higher retention time as compared with monomers of the excess enantiomer.9–13 Since the separation is considered to be based on the internal chirality of enantiomerically enriched compounds it cannot be applied for resolution of racemates.
Some of the pioneering reports about separation of enantiomers under achiral conditions: most of the reports on separation of enantiomers under achiral conditions have been covered and discussed in the reviews mentioned above.1,13 Nevertheless, it may be pertinent to cite some of the pioneering work in the area of separation of enantiomers from non racemic mixtures under achiral conditions. These can be categorized as (i) experiments reporting separation of certain non-racemic mixtures by achiral phase chromatography or other methods (e.g., distillation, sublimation), (ii) theoretical models to explain enantioselective effects in achiral phase chromatography (applicable to other methods also), and (iii) reviews covering these aspects.
Cundy and Crooks14 performed HPLC (on Partisil-ODS) experiments with samples of racemic 14C-labelled nicotine co-injected with unlabelled (S)-(−)-nicotine or its antipode as standard of varying enantiomeric composition, the 14C activity was found to have divided into two peaks which demonstrated that the two 14C peaks observed were due to the separated enantiomers of the radiolabelled material. It was contended that this separation of enantiomers in achiral environment was due to diastereomeric interaction of enantiomers (of nicotine) that existed predominantly as an association between two like enantiomers which produced a dimer with slightly different properties in comparison to the one composed of unlike enantiomers (the diastereomeric forms). It was considered that hydrogen bonding was playing a significant role as nicotine was predominantly protonated on the l′-N atom, at the pH (6.8) used in the study, and saturation resulted from solvation effects; small amounts of hetero-dimer had a reduced affinity for a mobile phase which was predominantly composed of solvated homo-dimers. This reduced affinity might have overcome when there was sufficient heterodimeric material present to promote its own salvation. The thermodynamics of these interactions may or may not be the same, but the proportion of homo- or hetero-dimers would certainly be under statistical control. The phenomenon responsible for this separation was not established as there was no evidence to suggest that nicotine associated in solution, but a concept of differential enantiomeric association between like and unlike optical isomers was provided.
Dobashi et al. got two split zones15 differing in enantiomer excess, in non-aqueous silica gel achiral HPLC (at a flow rate of 1 mL min−1 and by UV detection at 230 nm) from enantiomerically enriched mixtures of chiral solute, N-acetylvaline tert-butyl ester. Hydrogen-bond association of the chiral solute brought about a self-induced enrichment of enantiomers in the migrating solute zone; binary associations were considered to occur through bidentate NH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (ester) hydrogen bonds in relatively nonpolar co-solvents, such as CCl4 and CDCl3. The separation was concluded to arise from differences in stability between diastereomeric dimers in the mobile phase process; they used the term autoseparation.
C (ester) hydrogen bonds in relatively nonpolar co-solvents, such as CCl4 and CDCl3. The separation was concluded to arise from differences in stability between diastereomeric dimers in the mobile phase process; they used the term autoseparation.
Collet16 explained that the enantiomers can be sorted out even by hand in a conglomerate (which is a mixture of (R) and (S) crystals in equal amounts, i.e., racemate); the very first resolution of any compound, i.e., racemic tartaric acid in the form of its sodium ammonium tartrate by Pasteur which also led him to the discovery of molecular chirality can be explained on this basis.
Gil-Av and Schurig17 proposed a theoretical model (improving upon the one proposed by Jung and Schurig18) taking into account the enantioselective effects (as per the few literature reports available till then) and suggested that such enantioresolution occurs when associations of the dimer type takes place between the molecules to be resolved; the model was based on the idea that owing to rapid and reversible association of monomers to dimers each molecule will change its partitioning behavior rapidly as function of time but cannot have a constant capacity factor. Gil-Av and Schurig17 did not determine the constants involved in their model and consequently did not know to what extent it could fit experiments. Trap and Schurig19 presented a theoretical model to simulate and verify unusual elution orders of enantiomers on an achiral stationary phase doped with a small amount of a chiral selector or achiral columns coupled with columns doped with a chiral selector and suggested that nonlinear effects caused by molecular association of enantiomers in non-racemic mixtures can cause unexpected effects in chiroptics, NMR spectroscopy, homogeneous catalysis, and chromatography.
Kagan and co-workers20 discovered and studied the first cases of nonlinear correlation between the enantiomeric excess of a chiral auxiliary and the enantiomeric yield of an asymmetric synthesis (in a stoichiometric or a catalytic mode) and showed in three experiments that there was a strong departure from the linear relationship usually assumed between the enantiomeric excess of a chiral auxiliary and the extent of the asymmetric synthesis; in order to check the absence of any nonlinear relationship it becomes necessary to use samples of chiral ligands with different enantiomeric excesses. Kagan et al.,21 observed an intriguing example where the minor enantiomer (S) was found in the first fraction of a sample enriched in the (R)-enantiomer. All the reports cited above, have discussed separation aspects related to non-racemic mixtures and not the issues related to resolution of racemic mixtures under both achiral phases in chromatography.
These reports leave behind certain scientific issues (as conceptual challenge) to be addressed, e.g., (i) is it possible to resolve a racemic mixture with both achiral phases in chromatography, (ii) how should the experiments be conducted to achieve such a resolution, and (iii) what would be the explanation.
Tateishi et al.,22 claimed a ‘conceptually new approach for the preparation of enantiomerically pure compounds from the racemates’ by achiral chromatography. In the experiment,22 an optically pure ‘chiral inducing reagent’ (CIR) and the rac-derivative were mixed (with a mole ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), and subsequently subjected to medium pressure liquid chromatography (MPLC) on an achiral column (10 μm of silica gel) using an achiral eluent (hexane–AcOEt = 1); there was no base line separation of enantiomers and, in terms of isolated yield, there was obtained only 21% or 26% of enantiomerically pure (S)-enantiomer from the starting racemic mixture when the enantiomeric composition was determined using chiral HPLC; they presumed a preferential formation of high-order hetero-association between (R)-enantiomer of the racemic substrate and (S)-enantiomer of an optically pure CIR which transformed the initial racemate into enantiomerically enriched fractions having different retention times.9–11
:1), and subsequently subjected to medium pressure liquid chromatography (MPLC) on an achiral column (10 μm of silica gel) using an achiral eluent (hexane–AcOEt = 1); there was no base line separation of enantiomers and, in terms of isolated yield, there was obtained only 21% or 26% of enantiomerically pure (S)-enantiomer from the starting racemic mixture when the enantiomeric composition was determined using chiral HPLC; they presumed a preferential formation of high-order hetero-association between (R)-enantiomer of the racemic substrate and (S)-enantiomer of an optically pure CIR which transformed the initial racemate into enantiomerically enriched fractions having different retention times.9–11
Though it was a report using achiral phase chromatography but the enantiomeric composition was determined using chiral HPLC. In our opinion application of chiral HPLC to verify/justify the resolution under achiral phase lost the usefulness of resolution under achiral phase.
Advantages of thin layer chromatography (TLC) comparing to high-performance liquid chromatography (HPLC) have been summarized by Sherma23 in a review of modern TLC in pharmaceutical and drug analysis. Some of the advantages of TLC include semi-preparative isolation of the separated compounds and photography of the chromatogram as a clearly visible evidence of separation.
The literature, as noted above, and the literature on impregnation of TLC plates, in different manners, by suitable chiral selectors for direct ‘resolution’ of a variety of compounds24–26 including certain β-blockers27–30 prompted us to mix the chiral selector and the racemate prior to application of sample on plain silica gel plates and to develop the chromatograms in solvent systems having no chiral additive so that both the phases at the time of chromatographic separation are achiral.
For this purpose, resolution of DL-selenomethionine (SeMet) (Fig. 1a) and three β-blockers, namely, atenolol (Atl), betaxolol (Bel) and orciprenaline (Orc); Fig. 1b–d) was attempted using (−)-quinine (Fig. 1e), a cinchona alkaloid and L-glutamic acid (L-Glu), an amino acid, as chiral inducing reagents (CIR) which are less expensive and easily available in chirally pure form. The chromatograms showing resolution were photographed. Experimental results have been supported (in terms of separation mechanism and elution order) theoretically by drawing the optimized structures of transient diastereomers based on density functional theory. To the best of authors' knowledge this is the first report on the resolution of these compounds with both achiral phases in TLC.
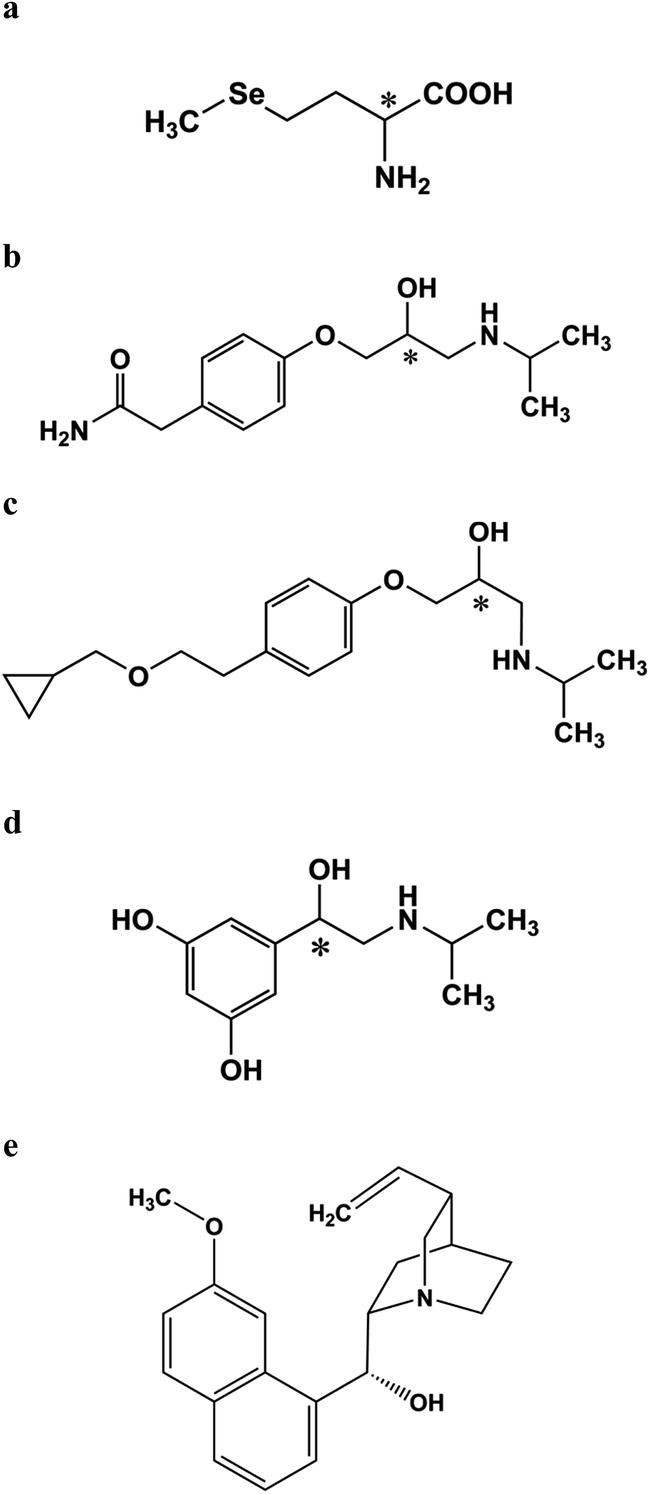 | ||
| Fig. 1 Structures of (a) Selenomethionine, (b) atenolol, (c) betaxolol, (d) orciprenaline (* represents chiral centre) and (e) (−)-quinine. | ||
Literature reports on biological/pharmaceutical importance and on enantioseparation of SeMet and the three β-blockers (by chiral liquid chromatography and by application of chiral derivatizing reagents) have not been described to keep the length of the paper short and to focus on enantioresolution of the analytes by TLC using both achiral phases.
2. Experimental
2.1 Chemicals and reagents
Reagent grade, L-(+)-SeMet (ee ≥ 98%; [α]25D = +18°, c = 2 in 2 M HCl; m.p. 256–257 °C) and racemic DL-(±)-SeMet (purity ≥ 99%) were purchased from Sigma-Aldrich (Bangalore, India). Enantiomerically pure glutamic acid (l-(+)-Glu, ee ≥ 99.5%, [α]25D +31.5 ± 1.0°, c = 5% in 5 M HCl, m.p. 185 °C) and, (S)-(−)-atenolol (assay 99%, [α]25D = −16°, c = 1 in 1 M HCl, m.p. 148–152 °C) and the racemic form (assay ≥ 98%, (±)-atenolol) were obtained from Sigma-Aldrich (St Louis, MO, USA). Betaxolol hydrochloride as Iobet eye drops (FDC Ltd. Bhiwadi, India) and orciprenaline sulphate as Alupent tablets (Zydus Healthcare, East Sikkim, India) were obtained from the local market. Sodium hydrogencarbonate (NaHCO3) and dichloromethane (CH2Cl2) of analytical reagent grade, acetonitrile (MeCN) and methanol (MeOH) of HPLC grade were obtained from E. Merck (Mumbai, India). Optically pure (−)-quinine (ee ≥ 98.0%, [α]25D = −165°, c = 2 in ethanol) and silica gel G with 13% calcium sulfate as binder, having lead, chloride and iron impurities up to 0.02%, with pH 7.0 in a 10% aqueous suspension was obtained from Merck (Mumbai, India). Other chemicals and reagents, of analytical grade, were obtained from BDH (Mumbai, India), Merck (Mumbai, India) and Sisco Research Laboratory (Mumbai, India).2.2 Equipment
Some of the equipments used were spectrophotometer (UV-1601, Shimadzu), a polarimeter (Krüss P3002, Germany) and pH meter (Cyberscan 510, Singapore). The Gaussian program 09 Rev. A.02 and hybrid density functional B3LYP with 6-31G* basis set were used to obtain optimized structures of [(−)–(+)] and [(+)–(+)]-diastereomers of β-blockers.2.3 Isolation of orciprenaline and betaxolol from commercial formulations
Isolation and purification of orciprenaline and betaxolol from their commercial formulations was carried out as per method described elsewhere. The recoveries of the titled compounds were of the order of 98% of the quantities reported on the commercial labels. The purity was confirmed by determining the melting point and UV absorption (λmax) which was in agreement with literature reports. These were used as racemic standards.2.4 Preparation of thin layer plates and solutions
TLC plates (10 × 5 cm with 0.5 mm thickness) were prepared by spreading the slurry of silica gel G (25 g in 50 mL water) with a Stahl-type applicator. The plates were activated by keeping overnight in oven (at 60 ± 2 °C). Besides, plain plates were prepared by adjusting the slurry at pH 4, 5, 8 and 9.Solutions (10−3 M) of (±)-SeMet and (+)-SeMet were prepared in 0.1 M NaHCO3, and solution of (−)-quinine (0.1%) was prepared by dissolving it in ethanol. Solutions of the three β-blockers (10−3 M) were prepared in methanol and solution of L-Glu (0.1%) was prepared in purified water. All solutions were filtered through a 0.45 μm filter.
2.5 TLC and isolation of enantiomers
Solutions of DL-(±)-SeMet and (−)-quinine were mixed in a mole ratio of (1:1). Solutions of each of the β-blockers (10−3 M) were also mixed with L-Glu in a mole ratio of (1:1). Solution of (+)-SeMet was mixed with that of (−)-quinine (in a mole ratio of 1:1). These were spotted (10 μL) on plain plates (having no chiral impregnating reagent in the slurry) using Hamilton syringe, 2 cm above the margin. Chromatograms were developed, with solvent systems having no chiral additive, using cleaned, dried and paper-lined rectangular glass chamber that was pre-equilibrated with the same solvent for 15 min at different temperatures (15 ± 2, 25 ± 2, and 35 ± 2 °C). The chromatograms were dried in air at room temperature for 10 to 15 min. The spots were located separately by (i) using iodine vapors as dark brown spots, and (ii) by spraying ninhydrin solution (0.3 g ninhydrin dissolved in 100 mL n-BuOH containing 3 mL CH3COOH) which was followed by heating of chromatogram at 70 °C for 10 min.
The spots representing enantiomers of SeMet (located by exposure to iodine vapours) were marked and iodine was allowed to evaporate off. Silica gel of the spots was scraped from nearly 30 chromatograms and the silica gel of corresponding spots so collected was extracted with water and the combined extract pertaining to each of the enantiomers of SeMet was filtered and concentrated under vacuum. Similarly, the separated enantiomers of β-blockers were extracted with methanol.
3. Results and discussion
3.1 Resolution
Different solvents, acetonitrile, methanol, water and dichloromethane and their mixtures (binary, ternary and quaternary) in various ratio were tried as solvent systems systematically to achieve resolution. The successful mobile phase was, acetonitrile–methanol–dichloromethane–water in the ratio of (10:1.5:0.5:1.5, v/v) and (6:3:3:1.5, v/v), respectively, for resolution of DL-(±)-SeMet and (RS)-(±)-β-blockers; chromatograms were developed in 10 min. hRF(RF × 100) values for all the analytes are shown in Table 1. Based on several TLC runs with different ratios of methanol and acetonitrile (along with other solvents) a high content of acetonitrile was found successful. Acetonitrile has a higher dielectric constant (37.5 D) and lower viscosity (0.343 cP at 25 °C) in comparison to methanol which has a lower dielectric constant (33 D) and higher viscosity (0.59 cP at 25 °C); as a result high content of methanol results into tailing and higher retention times.
| Analyte | CIR | Mobile Phase (v/v) | hR F pure | hR F from racemic mixture | R S | |
|---|---|---|---|---|---|---|
| (−) | (+) | (−) | ||||
| a R s, resolution; hRF = RF × 100. Approach (Pre-mixing of CIR with analyte); the CIR and the mobile phase was the same for resolution of three β-blockers. | ||||||
| Atenolol | L-Glu | CH3CN–CH3OH–CH2Cl2–H2O (6:3:3:1.5) |
28 | 44 | 28 | 1.7 |
| Orciprenaline | 33 | 53 | 33 | 2.1 | ||
| Betaxolol | 31 | 50 | 31 | 2.0 | ||
| SeMet | (−)-Quinine | CH3CN–CH3OH–CH2Cl2–H2O (10:1.5:0.5:1.5) |
13 | 26 | 13 | 1.69 |
Photograph of the actual chromatogram showing TLC resolution of (±)-SeMet is given as Fig. 2. Fig. 3 is the chromatogram for resolution of (+)-, and (−)-Atl in a ratio of 1:99 while Fig. 4 is the photograph of an actual chromatogram showing resolution of (±)-Orc, (±)-Bel and (±)-Atl from racemic mixture by TLC. Resolution (RS) was calculated by dividing the distance between two spots by the sum of two spot radii; a value of 1.50 was taken as indicative of complete resolution whereas a value of 1.20 or below indicated incomplete resolution;31 the highest (RS) was calculated to be 2.1 for enantiomers of Orc, and 1.6 for enantiomers of SeMet, in this study.
 | ||
| Fig. 2 Photograph of an actual chromatogram showing resolution of (±)-SeMet. From left to right: Line 1, lower spot is of (−)-isomer and the upper spot is of (+)-isomer (resolved from the racemic mixture); Line 2, pure (+)-isomer. Mobile phase: CH3CN–CH3OH–CH2Cl2–H2O (10:1.5:0.5:1.5, v/v). Solvent front: 8 cm; temperature: 25 ± 2 °C; detection: ninhydrin solution (0.3% in n-BuOH); development time: 10 min. | ||
 | ||
| Fig. 3 Photograph of an actual chromatogram showing resolution of (±)-Atl. From left to right: Line 1, (+:−), 1:99, lower spot for (−)-isomer and upper spot for (+)-isomer; line 2, is of (−)-isomer. Mobile phase: CH3CN–CH3OH–CH2Cl2–H2O (6:3:3:1.5, v/v). Solvent front: 8 cm; temperature: 25 ± 2 °C; detection: iodine vapor; development time: 10 min. | ||
 | ||
| Fig. 4 Photograph of an actual chromatogram showing resolution of (±)-Atl, (±)-Orc and (±)-Bel. From left to right: Line 1, 2 and 3, respectively, for (±)-Orc, (±)-Bel and (±)-Atl, lower spot is of (−)-isomer, and the upper spot is of (+)-isomer (resolved from the racemic mixture). Mobile phase: CH3CN–CH3OH–CH2Cl2–H2O (6:3:3:1.5, v/v). Solvent front: 8 cm; temperature: 25 ± 2 °C; detection: iodine vapor; development time: 10 min. | ||
To compare and verify the migration order of the isomers getting separated from racemic mixture, pure enantiomer was applied in parallel on the TLC plates. It was found that the migration distance of (+)-SeMet was more than that of (−)-isomer. In case of β-blockers the lower spot was of (−)-enantiomer and the migration distance of (+)-enantiomer was more than that of (−)-enantiomer.
Since (−)-quinine is insoluble in cold water only enantiomers of SeMet, from the two cut spots, went into solution. The optical purity of separated enantiomers was examined by polarimeter and it was found in agreement with the literature report. The specific rotation of the isolated (+)-SeMet was found to be (+) 17.90° which was in agreement with literature (specific rotation for (+)-SeMet is (+) 18.0°, c = 2, 2 M HCl).32 Similarly, the separated enantiomers pertaining to (+)- or (−)-isomers of the said β-blockers went into methanol as L-Glu is insoluble in methanol. The concentration for each isomer was estimated by using ‘calibration plots’. Determination of optical rotation was carried out using polarimeter. Specific rotation for lower spot of Atl was found to be −10.74° (c = 0.5, methanol) which was in agreement with the literature reports.32
In the present studies, racemic mixture of SeMet was pre-mixed with (−)-quinine, as the CIR, before applying on plates. There occurred resolution and the two enantiomers were recovered in 1:1 ratio (established by polarimetric studies). Thus the basis of resolution is considered to be the same as explained13,22 where ‘preferential formation of high-order (enantiomeric) associates’ is envisaged. Instead of ‘mixed homo-/heterochiral high-order species’ with different retention times there occurred formation of diastereomeric associates, of the type [(−)-SeMet–(−)-quinine] and [(+)-SeMet–(−)-quinine], leading to transformation of the initial racemate into mixture of diastereomers with different chromatographic mobilities in achiral phases and hence resolution. The same explanation holds good for enantioresolution of β-blockers under achiral phase chromatographic conditions.
Since the formation of diastereomeric associates, of the type [(−)-SeMet–(−)-quinine] and [(+)-SeMet–(−)-quinine], has been considered and the native enantiomer has been isolated from such a diastereomer after extracting the latter from the silica gel cut from the chromatograms it can be contended that the retention factor for the additive is the same as it is for the enantiomer of the analyte. The difference in the retention factors is for the diastereomers.
Isolation of two enantiomers from TLC plates confirmed the formation of diastereomers via non-covalent reversible interactions. Presence of the CIR in both the spots, resolved under achiral phase chromatographic conditions, was evidenced when the residual silica gel (remaining after extracting enantiomers of SeMet or β-blockers under study) tested positive for the presence of (−)-quinine or amino acid. Further evidence for formation of such diastereomers was obtained when the chromatogram was sprayed with ninhydrin and each spot was visible in characteristic pink-purple colour for the presence of SeMet. However, there appeared a light pink background on the plate (impregnated with L-Glu) but the resolved spots were visible with more intensity and sharpness.
Among several possible combinations, using different ratio of CIR a ratio of 1:1 [(±)-SeMet:(−)-quinine] gave the best result because this proved to be the required ratio for the formation of diastereomers. The same ratio was found successful for formation of diastereomers of β-blockers. The effect of temperature and pH was investigated on the formation of diastereomers (formed through noncovalent interactions); the best results for enantioresolution of DL-SeMet were obtained at 25 °C and pH 8, while in case of β-blockers a temperature of 25 °C and pH 5 were found as optimized conditions.
This approach was found to be successful for direct and sensitive resolution of DL-SeMet and β-blockers from racemic mixture under achiral phases of chromatography and for detection of their enantiomers in a range lower than the limits prescribed (1%) for pharmaceuticals in industry. This approach was also found successful to separate non-racemic mixture of (+)- and (−)-enantiomers of β-blockers in the ratio of 1:99; Fig. 3 shows a representative chromatogram. It may be considered better and a novel approach in comparison to the method reported by Tateishi et al.,22 which indeed is another approach to enantiomeric enrichment rather than resolution of racemic mixture. Nevertheless, the present approach is successful in resolving racemic mixture under achiral phases of chromatography but has the difficulty in getting quantitative yields.
3.2 Support to the proposed mechanism and elution order
Lowest energy structures of transient diastereomers of Orc, formed with L-(+)-Glu, have been developed by DFT using Gaussian 09 Rev. A.02 program and B3LYP hybrid density functional with 6-31G* basis set to support the explanation for their formation and separation.In [(−)-(+)]-diastereomer, –COO− group present on stereogenic centre C1 of (+)-Glu and NH2+ group (in –CH2NHCH(CH3)2 moiety) present on stereogenic centre C2 of (−)-Orc, are oriented closely in space and provide a strong electrostatic interaction. The structure (in Fig. 5a) shows that (i) amino group (–NH3+) on stereogenic centre C1 of (+)-Glu and –OH on stereogenic centre C2 of (−)-Orc, and (ii) carboxyl group (–COO−) of (+)-Glu and –OH group present on benzene ring of Orc are in linear positions (and in the same plane) and provide strong H-bonding. Besides, there may be van der Waal's forces, steric, and hydrophobic interactions and other forms of electron donation and acceptance that are readily reversible and are responsible for formation and separation of transient diastereomers (to support three point interaction rule) which cannot be depicted in this figure (Fig. 5a).
 | ||
| Fig. 5 Structures of transient diastereomers of (−)-Orc with (+)-Glu, optimized and drawn using the Gaussian 09 Rev. A.02 program and B3LYP hybrid density functional with 6-31G* basis set. (a), [(−)–(+)]-diastereomer, and (b), [(+)–(+)]-diastereomer; explanation described under “support to the proposed mechanism via developing optimized structures for the transient diastereomers using DFT”. | ||
In [(+)–(+)]-diastereomer (Fig. 5b), the –COO− group on C1 of (+)-Glu and NH2+ group (in, –CH2NHCH(CH3)2 moiety) on C2 of (−)-Orc appear to be away from each other. Similarly, –NH3+ group on C1 of (+)-Glu and –OH present on C2 of (−)-Orc are also not seen in the same plane. Thus, there are weaker interactions as compared to those seen in [(−)–(+)]-diastereomer. However, carboxyl group (–COO−) of (+)-Glu and –OH of benzene ring in Orc appear to be in the same plane and show H-bonding.
Further, planarity and more number of bonds contribute to the enhanced hydrophobicity leading to increased retention time and lower RF for [(−)–(+)]-diastereomer than the other diastereomer. The results clearly show that, RF of [(+)–(+)]-diastereomer of each of the three β-blockers is greater than the RF of corresponding [(−)–(+)]-diastereomer.
In the present case, it can thus be argued that [(−)–(+)]-diastereomer represents hetero-chiral enantiomeric associate which is preferentially formed and is more stable9,12,13 due to linearity and strong interactions (Fig. 5a) in comparison to [(+)–(+)]-diastereomer (representing homo-chiral enantiomeric associate). Further, planarity and more number of bonds contribute to the enhanced hydrophobicity leading to increased retention time and lower RF for [(−)–(+)]-diastereomer than the other diastereomer. The results clearly show that, RF of [(+)–(+)]-diastereomer of each of the three β-blockers is greater than the RF of corresponding [(−)–(+)]-diastereomer.
As per Jung and Schurig18 model, (i) enantiomeric associates (as dimers) are assumed to be formed only in solution and not on the adsorbent and the self association in mobile phase caused separation by changing the relative time fractions spent by each enantiomer as the monomer if the enantiomeric associates (say, dimers) are of unequal stability, (ii) enantiomeric associates as dimers are assumed to be formed only on the adsorbent and not in mobile phase. In other words, it is assumed like an “adsorbed chiral layer” and the situation is compared to resemble the chromatography on chiral phases, where the capacity factors of the enantiomers are different owing to the stereoselective interactions with the chiral selector. Thus, in the present case too, it can be contented and explained in terms of self-association occurring on the stationary phase with consequent stereoselective interactions resulting into an excess of one enantiomer along with dissymmetry in both phases and, therefore, the stabilities of hetero- and homoenantiomeric associates (say, dimers) were unequal. The separation starts because of the creation of an anisometric medium and dissymmetric system by the presence of existing enantiomeric excess.
Chromatography of a non-racemic mixture of enantiomers, with achiral phases (and in the absence of any chiral inducing reagent), can furnish fractions which differ in ee; this achiral phase chromatography could be used to further enrich a sample in one enantiomer. Since ee effects can also occur during reactions with achiral reagents, further transformations of an enantiomer-enriched product may furnish false information on its ee. Therefore, it can be argued, by the same token, that chromatography could not generally be a safe method for purification of the product of an enantiomer-differentiating process, if only the ee of a purified portion of that product is taken as a measure of the efficiency of the process.
3.3 Method validation
Mean value (n = 5) of relative standard deviation (RSD) for precision was found in the range of 0.94 to 1.42 for both the separating enantiomers and recovery was within the range of 93.6 to 97.5%. Limit of detection was established by spiking different concentrations of DL-SeMet (in the range of 0.1–0.5%) with fixed amount of L-SeMet in quinine solution. These were spotted for TLC separation and detection. Detection of D-SeMet in the solution of L-SeMet was found up to 0.18%. Direct TLC method was found capable to detect 0.36 μg mL−1 (0.18 μg mL−1 of each enantiomer) of DL-SeMet and for β-blockers it was the range of 1.2–1.8 μg mL−1. Fig. 3 is showing a representative chromatogram of resolution of (+)- and (−)-Atl (in a ratio of 1:99).
4. Conclusion
The present approach has the following features, (i) resolution of a racemic and non-racemic mixture is possible with the help of a CIR using achiral phase chromatography, (ii) isolation of native enantiomer is possible, (iii) the approach is expected to be of general application for obtaining enantiomerically pure samples, (iv) it is overall simple, less expensive and does not require analysis/determination of enantiomeric composition by chiral HPLC, and (v) formation of diastereomers was supported by isolation of pure enantiomers and theoretically by DFT. TLC has also been used as an additional characterization technique in assisting analysis of the fast and slow competing enantioselective conversion reactions to determine absolute configuration of secondary alcohols in micromole quantities33 and the present approach opens another area for general application and utility of TLC.Acknowledgements
Authors are grateful to the Council of Scientific Industrial Research (CSIR), New Delhi, India, for awarding a senior research fellowship to HN, and to the AvH-Stiftung, Bonn, Germany for award of research fellowship (to R.B.).References
- IUPAC, Compendium of Chemical Terminology the “Gold Book”, ed. A.D.McNaught and W.Wilikinson, Blackwell Scientific Publications, Oxford, 2nd edn, 1997; PAC, 1996, 68, 2193; Basic terminology of stereochemistry; IUPAC Recommendations 1996, page 2217).
- J. Martens and R. Bhushan, J. Liq. Chromatogr., 1992, 15, 1–27 CrossRef CAS.
- R. Stephani and V. Cesare, J. Chromatogr. A, 1998, 813, 79–84 CrossRef CAS.
- R. M. Nicoud, J. N. Jaubert, I. Rupprecht and J. Kinkel, Chirality, 1996, 8, 234–243 CrossRef CAS.
- R. Baciocchi, G. Zenoni, M. Mazzotti and M. Morbidelli, J. Chromatogr. A, 2002, 944, 225–240 CrossRef CAS.
- S. Ogawa, T. Nishimine, E. Tokunaga, S. Nakamura and N. Shibata, J. Fluorine Chem., 2010, 131, 521–524 CrossRef CAS.
- V. J. Mayani, S. H. R. Abdi, R. I. Kureshy, N. H. Khan, S. Agrawal and R. V. Jasra, Chirality, 2009, 21, 255–261 CrossRef CAS PubMed.
- V. J. Mayani, S. H. R. Abdi, S. V. Mayani, H. C. Kim and S. K. Park, Chirality, 2011, 23, 300–306 CrossRef CAS.
- T. Katagiri, C. Yoda, K. Furuhashi, K. Ueki and T. Kubota, Chem. Lett., 1996, 2, 115–116 CrossRef.
- V. A. Soloshonok, Angew. Chem., Int. Ed., 2006, 45, 766–769 CrossRef CAS PubMed.
- V. A. Soloshonok and D. O. Berbasov, J. Fluorine Chem., 2006, 127, 597–603 CrossRef CAS.
- V. A. Soloshonok and D. O. Berbasov, Chim. Oggi, 2006, 24, 44–47 Search PubMed.
- J. Martens and R. Bhushan, Helv. Chim. Acta, 2014, 97, 161–187 CrossRef CAS.
- K. C. Cundy and P. A. Crooks, J. Chromatogr., 1983, 281, 17–33 CrossRef CAS.
- A. Dobashi, Y. Motoyama, K. Kinoshita, S. Hara and N. Fukasaku, Anal. Chem., 1987, 59, 2209–2211 CrossRef CAS.
- A. Collet, in Optical resolutions by crystallization methods, Chiral Separations by HPLC, ed. Krstulovic, A. M., Ellis Horwood Ltd., Chichester, 1989, pp. 81–84 Search PubMed.
- E. Gil-Av and V. Schurig, J. Chromatogr. A, 1994, 666, 519–525 CrossRef CAS.
- M. Jung and V. Schurig, J. Chromatogr., 1992, 605, 161–166 CrossRef CAS.
- O. Trapp and V. Schurig, Tetrahedron: Asymmetry, 2010, 21, 1334–1340 CrossRef CAS.
- C. Puchot, O. Samuel, E. Duiiach, S. Zhao, C. Agami and H. B. Kagan, J. Am. Chem. Soc., 1986, 108, 2353–2357 CrossRef CAS PubMed.
- P. Diter, S. Taudien, O. Samuel and H. B. Kagan, J. Org. Chem., 1994, 59, 370–373 CrossRef CAS.
- K. Tateishi, S. Tsukagoshi, T. Nakamura, S. Watanabe, V. A. Soloshonok and O. Kitagawa, Tetrahedron Lett., 2013, 54, 5220–5223 CrossRef CAS.
- J. Sherma, Pharmacopeial Forum, 2001, 27, 3420 Search PubMed.
- R. Bhushan and S. Dixit, Biomed. Chromatogr., 2012, 26, 962–971 CAS.
- R. Bhushan and J. Martens, Biomed. Chromatogr., 1997, 11, 280–285 CrossRef CAS PubMed.
- R. Bhushan and J. Martens, in Handbook of Thin Layer Chromatography, ed. J. Sherma and B. Fried, Marcel Dekker, New York, 3rd edn, 2003, pp. 373–415 Search PubMed.
- R. Bhushan and G. T. Thiong'o, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 1998, 708, 330–334 CrossRef CAS.
- R. Bhushan and C. Agarwal, Biomed. Chromatogr., 2008, 21, 129–134 CAS.
- R. Bhushan and S. Tanwar, Biomed. Chromatogr., 2008, 22, 1028–1034 CrossRef CAS PubMed.
- R. Bhushan and S. Tanwar, J. Chromatogr. A, 2010, 1217, 1395 CrossRef CAS PubMed.
- Handbook of TLC, ed. J. Sherma and B. Fried, Marcel- Dekker, New York, 1990, p. 50 Search PubMed.
- Sigma-aldrich.com, 2003; Sigma Aldrich Catalogue, 2013.
- A. J. Wagner and S. D. Rychnovsky, J. Org. Chem., 2013, 78, 4594–4598 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |