
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
D-Glucose based syntheses of β-hydroxy derivatives of L-glutamic acid, L-glutamine, L-proline and a dihydroxy pyrrolidine alkaloid†
K. S. Ajish
Kumar
* and
Subrata
Chattopadhyay
Bio-organic Division, Bhabha Atomic Research Centre, Trombay, Mumbai-400085, India. E-mail: ajish@barc.gov.in
First published on 16th February 2015
Abstract
The β-hydroxy derivatives of L-glutamic acid, L-glutamine and L-proline, useful for peptide/protein studies, were synthesized starting from D-glucose. The C2 carbon in D-glucose provided the carboxylic acid functionality, while the amino and β-hydroxy groups of the amino acids were amenable from the C3 and C4 hydroxy groups of the sugar, respectively. The key intermediate with appropriate carbon framework of the target molecules was constructed by homologation of a suitable azido-D-glucofuranose derivative using the Arndt–Eistert reaction.
Introduction
Amino acids and monosaccharides constitute the major building blocks of the complex molecular systems that are vital for life. There has been a constant effort to understand the structures and functions of such systems by synthesizing them either chemically or biologically.1 Hence the importance of modified amino acids as ligating agents, in the synthesis of natural proteins has received considerable attention over the years.2 These amino acids are also the key ingredients for the synthesis of modified proteins, that help in understanding the structure–activity relationship,3 and lantibiotic4a,b study of the peptides of interest, besides providing peptidomimetic drugs.4c Native chemical ligation (NCL) forms the basis of modern chemical synthesis of native, modified or cyclic peptides and proteins of moderate sizes,5 and is extensively used to synthesize complex protein targets.2,6Although the NCL approach has enriched peptide ligation chemistry, the required thiol/selenol-containing amino acids, which are essential, are accessible only through lengthy syntheses. The nonproteinogenic amino acids, possessing suitably placed (at the β/γ-position) hydroxy group(s) along the side chain are useful precursors of the corresponding thiol and selenol derivatives, required for NCL. A few commercially available hydroxy derivatives of natural amino acids, such as, β-hydroxy phenylalanine, β-hydroxy valine, and β-hydroxy leucine have been transformed to the corresponding thiol intermediates and used in NCL.7 Meanwhile, the β/γ-hydroxy derivatives of glutamic acid, glutamine, lysine, arginine, and aspartic acid have also been synthesized in different laboratories, and their mercapto derivatives are proven residues for the assembly of peptides using NCL.8 Moreover, many of these hydroxy amino acids are constituents of several natural products with intrinsic biological function.9 Overall, both as unnatural building blocks and target compounds, the β/γ-hydroxylated amino acids are attractive synthetic targets. Consequently, several target-specific10 as well as multi-target oriented11 syntheses of the hydroxy amino acids have been reported.
Designing a common strategy, for various bioactive molecules has vital significance in organic synthesis. This can provide an economically accessible pathway to an array of discrete compounds from a single starting molecule.12 The natural amino acids glutamic acid (Glu), glutamine (Gln) and proline (Pro) possess a similar five-carbon skeleton. It was hypothesized that the synthesis of suitable hydroxy derivatives of these may be realized using a common strategy. Hence, in view of our interest in modified amino acid synthesis, applicable for protein synthesis and study,13a–c we formulated a general strategy for synthesizing the β-hydroxy derivatives of Glu (1a), Gln (1b) and Pro (1c) starting from inexpensive D-glucose. The corresponding β-hydroxy azido acids were also synthesized as the masked amino acids, because similar compounds are proven candidates for Staudinger ligation in peptides/proteins syntheses.14 In addition, several derivatives of 1a–1c, possessing different orthogonal ester protections (Me/allyl/benzyl) were synthesized so that they can be converted to free acids under different reaction conditions. Finally, in view of our interests on iminosugars,13d,e we have transformed one of the intermediates into a biologically important pyrrolidine alkaloid 2. The chemical structures of the target compounds are shown in Fig. 1. Amongst the chosen targets, L-glutamate is an important nutrient in biochemical pathways like gluconeogenesis and ammonia detoxification,15a and also plays a major role in learning, memory and neuronal development in mammalian central nervous system.15b,c
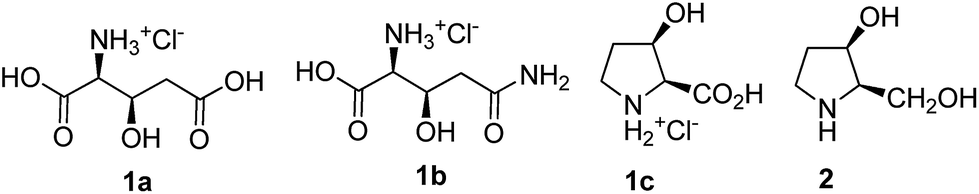 | ||
| Fig. 1 Chemical structures of the synthesized compounds. | ||
Results and discussion
In the retrosynthetic analysis, we conceived that the C2 carbon in D-glucose would furnish the carboxylic acid functionality, while the C3 and C4 hydroxy groups would provide the required amino/azido and β-hydroxy groups, respectively, of the targeted β-hydroxy amino acid derivatives. The synthesis commenced with the known D-glucose-derived azido aldehyde 3,13e,16 which was subjected to Pinnick oxidation (NaClO2/NaH2PO4/30% H2O2)17 to furnish the azido acid 4 in 91% yield. The acid 4 was activated as a mixed anhydride using ethyl chloroformate, and subsequently reacted with CH2N2 in Et2O to give the α-diazo ketone 5 in 80% yield. Wolff rearrangement18 of 5 in the presence of PhCO2Ag and Et3N in MeOH afforded the homologated methyl ester 6 (55%) that served as the common intermediate for all the target amino acid derivatives.As the first application of 6, we attempted its conversion to the β-hydroxy glutamic acid derivatives. To this end, its 1,2-acetonide group was deprotected using aqueous trifluoroacetic acid (TFA), and the resultant hemiacetal was subjected to cleavage with NaIO4 to yield the intermediate azido aldehyde. This on Pinnick oxidation afforded the glutamic acid derivative 7, containing a formylated C-3 hydroxy group (79%, over three steps). The formyl group in 7 could be selectively de-masked with aqueous saturated NaHCO3 in THF to obtain the hydroxy acid 8 (89%). Compound 7 was also transformed to a fully masked Glu derivative 9 (84%) by reacting with allyl bromide in the presence of NaHCO3 in anhydrous DMF. As above, the formyl group in 9 could be selectively removed with NaHCO3 in THF at room temperature to obtain the β-hydroxy diester 10 in 87% yield.
Amino acid 10 is not suitable for Fmoc-SPPS as the side chain methyl ester is not easily cleavable under acidic/reduction condition. Hence it was thought of synthesizing a benzyl ester derivative 14 which would also serve as an ideal starting material for the various C3-substituted glutamic acid derivatives. For this, the ester function in 6 was hydrolyzed using LiOH in aqueous THF to afford the carboxylic acid 11 (86%), which on treatment with benzyl chloroformate (CbzCl) in the presence of Et3N and 4-dimethylaminopyridine (DMAP) afforded the benzyl ester 12 in 63% yield. The ester 12 was directly transformed to the hydroxy azido acid 13 by a one-pot four-steps reaction sequence. Thus, acidic hydrolysis of the 1,2-acetonide function of 12, NaIO4 cleavage of the resultant diol to the intermediate aldehyde, followed by Pinnick oxidation and alkaline hydrolysis furnished the desired C-3 hydroxy acid 13 in 71% yield. This was esterified with allyl bromide and NaHCO3 to afford another glutamic acid precursor 14 in 87% yield. Compound 14 is a template on which all the functionalities except the β-hydroxy group is protected and thus is a suitable precursor for the synthesis of C-3 substituted glutamic acid derivatives, e.g. β-mercapto glutamic acid. Such a transformation has already been established from similar hydroxy derivatives of various amino acids.7,8 Next, to confirm the stereochemistry at α and β-carbon in 14 it is necessary to convert it to a known derivative of β-hydroxy glutamic acid. For this azido acid 13 was opted as suitable substrate thus, a one pot reduction of azide functionality and debenzylation of ester using 10% Pd/C in MeOH–HCl afforded the fully unmasked β-hydroxy glutamic acid 1a in 95% yield (Scheme 1). The spectral and analytical data of 1a wherein agreement with that reported.10q
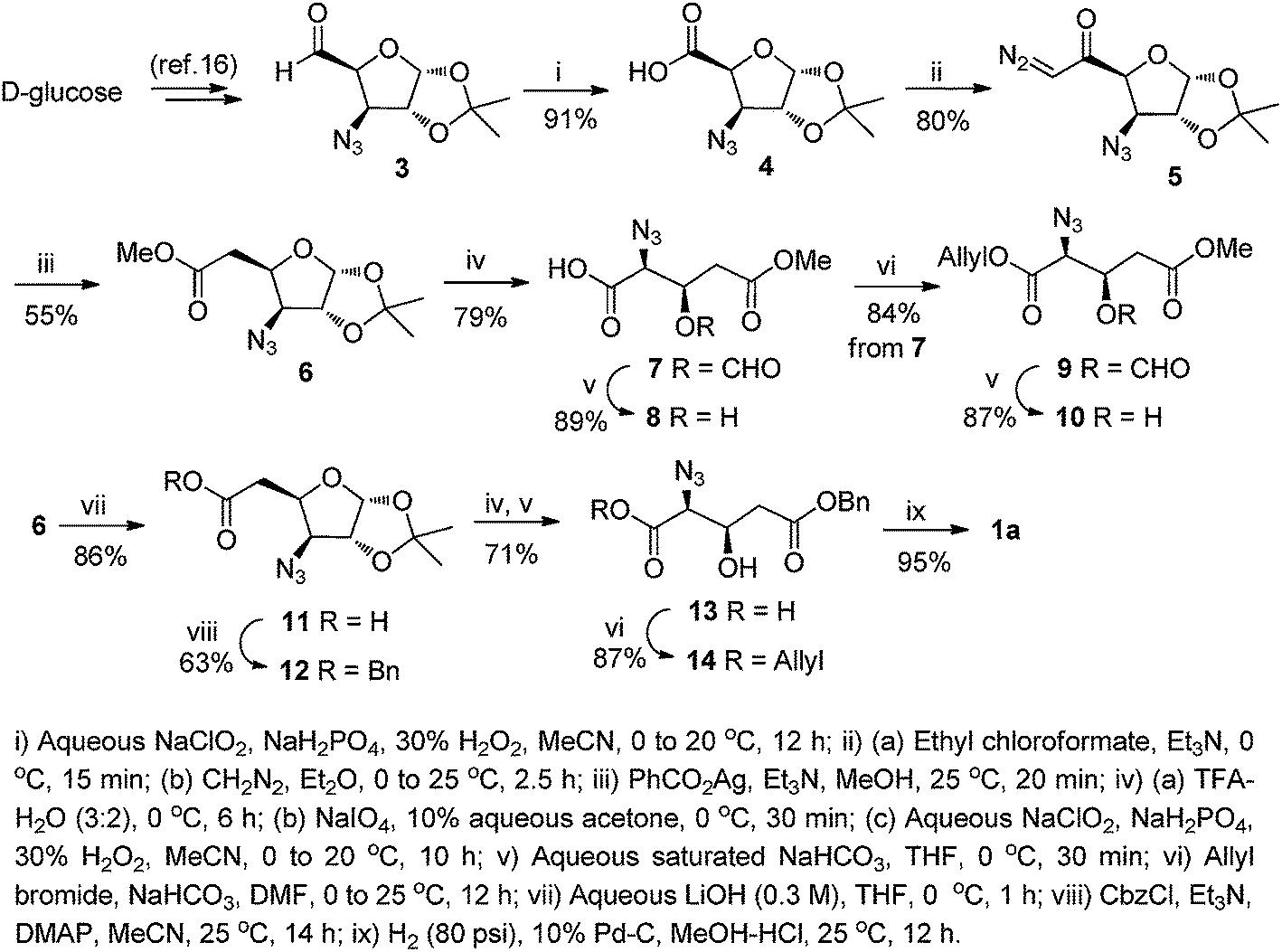 | ||
| Scheme 1 Synthesis of β-hydroxy glutamic acid derivatives. | ||
For the synthesis of the β-hydroxy glutamine 1b, the benzyl amide of compound 11 was envisaged to serve as the masked amino equivalent of glutamine. Hence, compound 11 was coupled with benzylamine using HBTU and HOBt in the presence of diisopropylethylamine (DIEA) in DMF to afford the desired amide 15 (62%). This was transformed to the acid 16 (72%, over 4 steps), following the same sequence of reactions used to transform 12 to 13. The acid 16 was converted to the N-benzyl azido analogue of β-hydroxy glutamine ester 17 (79%) by a base-catalyzed reaction with allyl bromide. However, catalytic hydrogenation of 16 over 10% Pd–C in MeOH even under a pressurized (80 psi H2) condition led to reduction of the azide functionality only, and furnished the hydrochloride of β-hydroxy glutamyl benzamide 18 instead of the fully unprotected β-hydroxy glutamine hydrochloride 1b. Our attempts to transform 16 to the desired product 1b with HCO2NH4/10% Pd–C/MeOH at room temperature as well as under reflux were also unsuccessful.
In an alternative method, the acid 11 was converted to the amide 19 (90%) with di-tert-butyl dicarbonate ((Boc)2O), (NH4)HCO3·NH2CO2NH4 and pyridine in MeCN. This was converted to the acid 20 (vide supra), which on catalytic hydrogenation afforded 1b in 90% yield. Azido acids, similar to 13, 16, and 20 are reported to be candidates for Staudinger ligation14 in peptide/protein synthesis (Scheme 2).
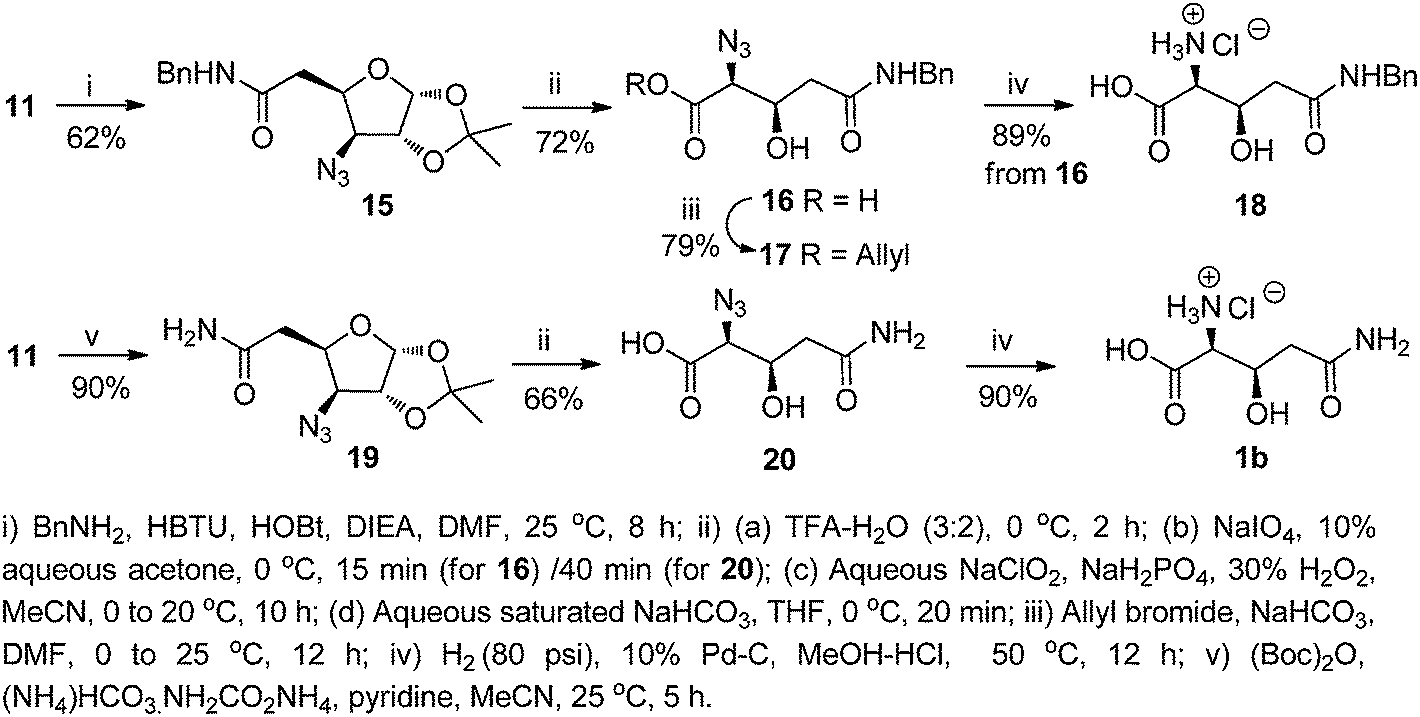 | ||
| Scheme 2 Synthesis of β-hydroxy glutamine derivatives. | ||
Next, we focused our attention to the synthesis of the β-hydroxy proline hydrochloride 1c and its derivative 23. It was also realized that the intermediates, generated in the process may be transformed to the pyrrolidine derivatives such as 2 that are of our own interest as bioactive iminosugars.13d,e In this direction, compound 6 was subjected to a catalytic transfer hydrogenation (HCO2NH4/10% Pd–C/MeOH) to afford the bicyclic lactam 21 in 91% yield via a tandem azide reduction and cyclization. The lactam 21 was reduced with LiAlH4 in THF under refluxing conditions, and the resultant amine functionality protected with CbzCl to furnish the N-Cbz protected bicyclic intermediate 22 in 57% yield (over two steps). The carbamate 22 was subsequently transformed to 1c as reported earlier.10i We also synthesized the allyl ester of β-hydroxy proline from 22 without any purification of the intermediates. For this, compound 22 was sequentially subjected to an acid-catalyzed ketal hydrolysis, NaIO4 cleavage, Pinnick oxidation and alkaline hydrolysis to obtain the crude acid. After drying in vacuo, the acid was subjected to a base-catalyzed allylation to furnish the β-hydroxy proline allyl ester 23 in 65% yield (over five steps). It is worth noting that the β-hydroxy esters 14, and 23 could also serve as precursors for functional group transformations at the free hydroxy group, because the subsequent deallylation can be accomplished under neutral and non-reducing conditions using a Pd(II) catalyst (Scheme 3).
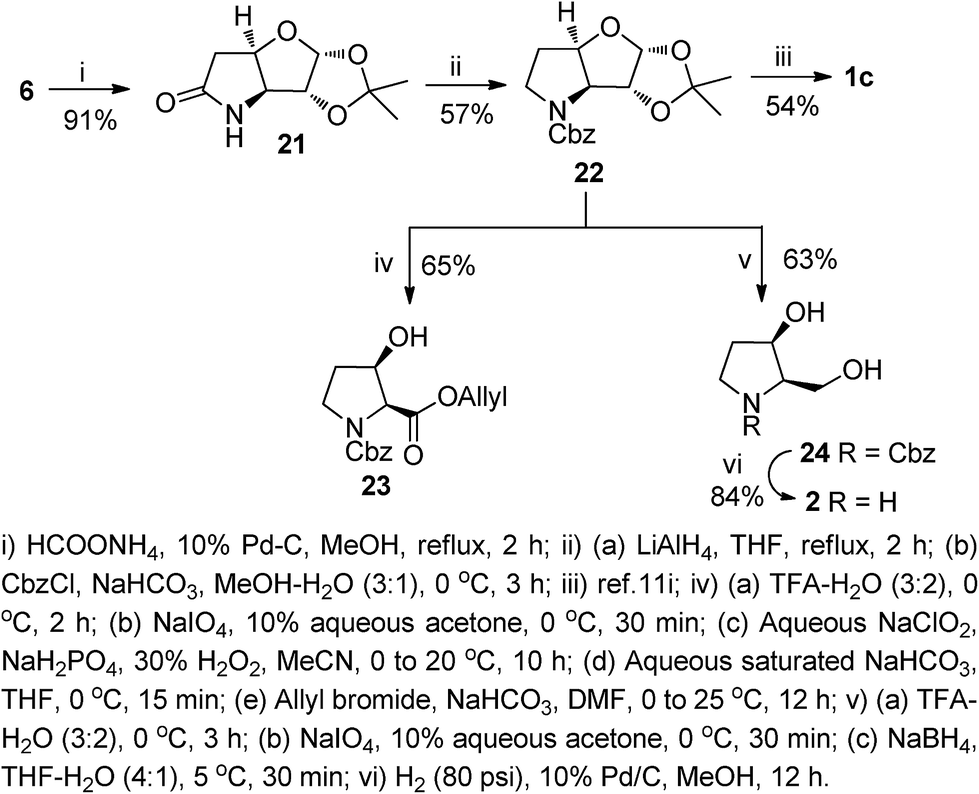 | ||
| Scheme 3 Synthesis of β-hydroxy proline derivatives and a pyrrolidine alkaloid. | ||
For the synthesis of the pyrrolidine iminosugar 2, the carbamate 22 was treated with aqueous TFA to unmask the acetonide group, and the resultant diol cleaved with NaIO4 to yield an aldehyde, which on NaBH4 reduction afforded the N-Cbz protected pyrrolidine 24. In the final step, the amino functionality in 24 was deprotected by catalytic hydrogenation over 10% Pd–C in MeOH to afford the desired dihydroxypyrrolidine 2 in 84% yield. Compound 2 is a versatile precursor for the 3,4-cis-substituted aza-sugars that show a wide range of biological activity. To our surprise unlike its enantiomer, only a few synthesis of 2 have been reported.19
Conclusions
In summary, we have devised an important strategy for the synthesis of β-hydroxy derivatives of glutamic acid, proline, glutamine, and a dihydroxy pyrrolidine alkaloid. Using this pathway different orthogonally protected hydroxy equivalent of glutamic acid, glutamine and proline are achievable. Noticeably, similar hydroxy amino acids with their functionalities protected as in 14, and 23 have been used for the synthesis of corresponding thiol derivatives and has been used for peptide ligation (NCL). Inexpensive reagents, cheap starting materials, and simple chemical transformations make this strategy a useful one for the synthesis of various protecting group variants of glutamine, glutamic acid and proline. Our efforts to transform the hydroxy derivatives to mercapto variants and their application in peptide synthesis are in progress and will be reported elsewhere.Experimental section
(3aR,5S,6R,6aR)-6-Azido-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid 4
To a stirred solution of 3 (3.31 g, 15.52 mmol) in MeCN (50 mL) were added NaH2PO4 (0.484 g, 3.10 mmol) in H2O (5 mL) and aqueous 30% H2O2 (2.3 mL, 17.1 mmol). The mixture was cooled to 0 °C, NaClO2 (2.24 g, 24.84 mmol) in H2O (6 mL) was dropwise added in 0.5 h and stirred at 20 °C till completion of the reaction (cf. 12 h, monitored by gas evolution). The reaction mixture was treated with sodium sulphate (1.00 g), and extracted with EtOAc (3 × 30 mL). Evaporation of solvent and column chromatography (silica gel, 10% MeOH/CHCl3) of the residue gave 4 (3.25 g, 91%) as a thick liquid. Rf = 0.30 (30% MeOH/CHCl3); [α]25D −31.3 (c 1.08, CHCl3); νmax/cm−1: 3430, 2108, 1683 cm−1; 1H NMR: δ 8.19 (broad s, D2O exchangeable, 1H), 6.01 (d, J = 3.4 Hz, 1H), 4.86 (d, J = 3.7 Hz, 1H), 4.67 (d, J = 3.4 Hz, 1H), 4.33 (d, J = 3.7 Hz, 1H), 1.48 (s, 3H), 1.32 (s, 3H); 13C NMR: δ 171.2, 113.1, 105.2, 82.9, 78.1, 66.5, 26.6, 26.2. Anal. calcd for C8H11N3O5: C, 41.92; H, 4.84; N, 18.33%. Found: C, 41.99; H, 4.90; N, 18.42%.3-Azido-6-diazo-3,6-dideoxy-1,2-O-isopropylidine-5-keto-α-D-xylo-1,4-furanose 5
To a cooled (0 °C) and stirred solution of 4 (3.12 g, 13.62 mmol) in THF (45 mL) was sequentially added Et3N (2.27 mL, 16.33 mmol) and ethyl chloroformate (1.43 mL, 14.97 mmol). After 15 min, the mixture was brought to room temperature and filtered through Celite-545. CH2N2 [prepared from N-nitrosomethyl urea (2.00 g, 19.40 mmol) and KOH (5 g)] in Et2O (50 mL) was dropwise added to the filtrate at 0 °C in 0.5 h. After stirring at room temperature for 2 h, the mixture was concentrated in vacuo, and the residue purified by column chromatography (silica gel, 10% EtOAc/hexane) gave 5 (2.78 g, 80%) as a thick liquid. Rf = 0.35 (20% EtOAc/hexane); [α]25D −90.3 (c 1.14, CHCl3); νmax/cm−1: 2105, 1720 cm−1; 1H NMR: δ 5.95 (d, J = 3.3 Hz, 1H), 5.81 (s, 1H), 4.71 (d, J = 3.1 Hz, 1H), 4.62 (d, J = 3.3 Hz, 1H), 4.35 (d, J = 3.1 Hz, 1H), 1.48 (s, 3H), 1.32 (s, 3H); 13C NMR: δ 191.1, 112.7, 105.1, 82.8, 82.5, 66.7, 54.7, 26.4, 26.0. Anal. calcd for C9H11N5O4: C, 42.69; H, 4.38; N, 27.66%. Found: C, 42.75; H, 4.44; N, 27.74%.Methyl[(3aR,5R,6S,6aR)-6-azido-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yfl]acetate 6
To a stirred solution of 5 (1.00 g, 3.95 mmol) in anhydrous MeOH (15 mL) was dropwise added silver benzoate (0.290 g, 1.26 mmol) in Et3N (3 mL). After stirring at 25 °C for 20 min, the mixture was concentrated in vacuo, and the residue purified by column chromatography (silica gel, 5% EtOAc/hexane) to obtain 6 (0.560 g, 55%) as a thick liquid. Rf = 0.52 (20% EtOAc/hexane); [α]25D −59.1 (c 1.17, CHCl3); νmax/cm−1: 2105, 1737, 1208 cm−1; 1H NMR: δ 5.81 (d, J = 3.7 Hz, 1H), 4.63 (d, J = 3.7 Hz, 1H), 4.60–4.44 (m, 1H), 4.08 (d, J = 3.2 Hz, 1H), 3.67 (s, 3H), 2.87–2.59 (m, 2H), 1.47 (s, 3H), 1.28 (s, 3H); 13C NMR: δ 170.6, 112.1, 104.1, 83.5, 75.4, 66.7, 51.9, 33.5, 26.5, 26.1; ESI-MS: calcd for [C10H15N3O5 + Na]+: 280.09 Da. Found: 279.88 Da. Anal. calcd for C10H15N3O5: C, 46.69; H, 5.88; N, 16.33%. Found: C, 46.67; H, 5.93; N, 16.43%.(2S,3R)-2-Azido-3-(formyloxy)-5-methoxy-5-oxopentanoic acid 7
A solution of 6 (0.702 g, 2.73 mmol) in TFA–H2O (3.00 mL, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) was stirred at 0 °C for 6 h. Azeotropic removal of TFA with toluene in vacuo afforded the intermediate hemiacetal (0.700 g, thick liquid), which was taken in acetone/water (10 mL, 9:1), cooled to 0 °C and NaIO4 (0.640 g, 2.99 mmol) added. After stirring for 0.5 h, the reaction mixture was concentrated in vacuo, the residue extracted with CHCl3 (3 × 10 mL), and the extract concentrated in vacuo to get the crude α-azido aldehyde (0.503 g, thick liquid). This was dissolved in MeCN (5 mL), treated successively with NaH2PO4 (0.08 g, 0.53 mmol) in H2O (1 mL) and 30% H2O2 (0.40 mL, 2.95 mmol), cooled to 0 °C, and NaClO2 (0.39 g, 4.36 mmol) in H2O (1.5 mL) added into it in 20 min. After stirring at 20 °C till completion of the reaction (∼10 h, monitored by gas evolution), the reaction mixture was treated with sodium sulphate (0.20 g), and extracted with EtOAc (3 × 15 mL). Concentration of the extract in vacuo followed by column chromatography (silica gel, 10% MeOH/CHCl3) of the residue gave 7 (0.500 g, 79% in three steps) as a thick liquid. Rf = 0.30 (30% MeOH/CHCl3); [α]25D −6.00 (c 1.0, CHCl3); νmax/cm−1: 2111, 1701 cm−1; 1H NMR: δ 9.31–8.71 (broad m, 1H, D2O exchangeable), 8.00 (s, 1H), 5.84–5.65 (m, 1H), 4.31 (d, J = 2.2 Hz, 1H), 3.68 (s, 3H), 2.84 (dd, J = 6.8, 1.2 Hz, 2H); 13C NMR: δ 171.6, 170.2, 159.9, 69.5, 62.3, 52.3, 35.2. Anal. calcd for C7H9N3O6: C, 36.37; H, 3.92; N, 18.18%. Found: C, 36.42; H, 3.98; N, 18.28%.
:2) was stirred at 0 °C for 6 h. Azeotropic removal of TFA with toluene in vacuo afforded the intermediate hemiacetal (0.700 g, thick liquid), which was taken in acetone/water (10 mL, 9:1), cooled to 0 °C and NaIO4 (0.640 g, 2.99 mmol) added. After stirring for 0.5 h, the reaction mixture was concentrated in vacuo, the residue extracted with CHCl3 (3 × 10 mL), and the extract concentrated in vacuo to get the crude α-azido aldehyde (0.503 g, thick liquid). This was dissolved in MeCN (5 mL), treated successively with NaH2PO4 (0.08 g, 0.53 mmol) in H2O (1 mL) and 30% H2O2 (0.40 mL, 2.95 mmol), cooled to 0 °C, and NaClO2 (0.39 g, 4.36 mmol) in H2O (1.5 mL) added into it in 20 min. After stirring at 20 °C till completion of the reaction (∼10 h, monitored by gas evolution), the reaction mixture was treated with sodium sulphate (0.20 g), and extracted with EtOAc (3 × 15 mL). Concentration of the extract in vacuo followed by column chromatography (silica gel, 10% MeOH/CHCl3) of the residue gave 7 (0.500 g, 79% in three steps) as a thick liquid. Rf = 0.30 (30% MeOH/CHCl3); [α]25D −6.00 (c 1.0, CHCl3); νmax/cm−1: 2111, 1701 cm−1; 1H NMR: δ 9.31–8.71 (broad m, 1H, D2O exchangeable), 8.00 (s, 1H), 5.84–5.65 (m, 1H), 4.31 (d, J = 2.2 Hz, 1H), 3.68 (s, 3H), 2.84 (dd, J = 6.8, 1.2 Hz, 2H); 13C NMR: δ 171.6, 170.2, 159.9, 69.5, 62.3, 52.3, 35.2. Anal. calcd for C7H9N3O6: C, 36.37; H, 3.92; N, 18.18%. Found: C, 36.42; H, 3.98; N, 18.28%.
(2S,3R)-2-Azido-3-hydroxy-5-methoxy-5-oxopentanoic acid 8
To a cooled (0 °C) and stirred solution of 7 (0.141 g, 0.61 mmol) in THF (3 mL) was added aqueous saturated NaHCO3 (1 mL). After stirring for 0.5 h, the reaction mixture was concentrated in vacuo, the residue acidified to pH 1 with aqueous 1 N HCl, and extracted with EtOAc (6 × 10 mL). The combined organic extracts were dried, concentrated in vacuo to obtain a residue, which on column chromatography (silica gel, 20% MeOH/CHCl3) gave 8 (0.110 g, 89%) as a thick liquid. Rf = 0.30 (30% MeOH/CHCl3); [α]25D −26.0 (c 1.10, CHCl3); νmax/cm−1: 3510, 2105, 1713, 1206 cm−1; 1H NMR: δ 6.64 (broad s, D2O exchangeable, 2H), 4.72–4.55 (m, 1H), 4.00 (d, J = 2.3 Hz, 1H), 3.71 (s, 3H), 2.78 (dd, J = 16.6, 8.3 Hz, 1H), 2.63 (dd, J = 16.6, 4.8 Hz, 1H); 13C NMR: δ 172.4, 68.7, 64.7, 52.3, 37.8. Anal. calcd for C6H9N3O5: C, 35.47; H, 4.47; N, 20.68%. Found: C, 35.44; H, 4.52; N, 20.77.5-Methyl 1-prop-2-en-1-yl(2S,3R)-2-azido-3-(formyloxy)pentanedioate 9
To a solution of 7 (0.500 g, 2.16 mmol) in DMF (3 mL) at 0 °C was added NaHCO3 (0.45 g, 5.40 mmol) followed by allyl bromide (0.23 mL, 2.70 mmol). The reaction mixture was stirred to 25 °C for 12 h, DMF was removed in vacuo, the residue extracted with EtOAc (3 × 10 mL), the organic extract dried and concentrated in vacuo. The product was purified by column chromatography (silica gel, 10% EtOAc/hexane) to afford 9 (0.493 g, 84%) as a viscous liquid. Rf = 0.40 (20% EtOAc/hexane); [α]25D −11.0 (c 1.16, CHCl3); νmax/cm−1: 3524, 2109, 1721, 1211 cm−1; 1H NMR: δ 7.96 (s, 1H), 6.00–5.70 (m, 2H), 5.43–5.21 (m, 2H), 4.69–4.63 (m, 2H), 4.23 (d, J = 2.9 Hz, 1H), 3.67 (s, 3H), 2.80 (d, J = 6.9 Hz, 2H); 13C NMR: δ 169.6, 167.2, 159.1, 130.8, 119.8, 69.3, 66.9, 62.3, 52.1, 35.1. Anal. calcd for C10H13N3O6: C, 44.28; H, 4.83; N, 15.49%. Found: C, 44.25; H, 4.80; N, 15.61%.5-Methyl 1-prop-2-en-1-yl(2S,3R)-2-azido-3-hydroxypentanedioate 10
Following the procedure used for 8, deformylation of 9 (0.230 g, 0.84 mmol) with aqueous saturated NaHCO3 (1.5 mL) in THF (5 mL) followed by usual work up and column chromatography (silica gel, 10% EtOAc/hexane) afforded 10 (0.180 g, 87%) as a thick liquid. Rf = 0.30 (20% EtOAc/hexane); [α]25D −17.0 (c 1.74, CHCl3); νmax/cm−1: 3611, 2102, 1716, 1202 cm−1; 1H NMR: δ 6.08–5.82 (m, 1H), 5.50–5.21 (m, 2H), 4.82–4.69 (m, 2H), 4.65–4.49 (m, 1H), 3.88 (d, J = 2.6 Hz, 1H), 3.71 (s, 3H), 2.76 (dd, J = 16.3, 7.6 Hz, 1H), 2.75–2.25 (broad s, D2O exchangeable, 1H), 2.58 (dd, J = 16.3, 4.5 Hz, 1H); 13C NMR: δ 172.1, 168.3, 131.0, 119.5, 68.9, 66.7, 64.9, 52.1, 37.8. Anal. calcd for C9H13N3O5: C, 44.44; H, 5.39; N, 17.28%. Found: C, 44.51; H, 5.46; N, 17.36%.[(3aR,5R,6S,6aR)-6-Azido-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl]acetic acid 11
To a cooled (0 °C) solution of 6 (1.20 g, 4.66 mmol) in THF (40 mL) was dropwise added an aqueous 0.3 M solution of LiOH (0.58 g, 13.99 mmol) over 30 min. After completion of reaction (cf. TLC, 30 min), the pH of the mixture was adjusted to 5–6 with aqueous saturated citric acid (20 mL), and extracted with EtOAc (3 × 30 mL). The combined organic extracts were dried, concentrated, and the residue purified by column chromatography (silica gel, 10% MeOH/CHCl3) to give 11 (0.980 g, 86%) as a thick liquid. Rf = 0.30 (30% MeOH/CHCl3); [α]25D −31.4 (c 1.0, CHCl3); νmax/cm−1: 3584, 2102, 1742 cm−1; 1H NMR: δ 9.00–7.81 (broad s, D2O exchangeable, 1H), 5.86 (d, J = 3.7 Hz, 1H), 4.68 (d, J = 3.7 Hz, 1H), 4.62–4.50 (m, 1H), 4.10 (d, J = 3.2 Hz, 1H), 2.94–2.65 (m, 2H), 1.51 (s, 3H), 1.31 (s, 3H); 13C NMR: δ 175.8, 112.3, 104.2, 83.5, 75.1, 66.7, 33.6, 26.5, 26.2. Anal. calcd for C9H13N3O5: C, 44.44; H, 5.39; N, 17.28%. Found: C, 44.40; H, 5.41; N, 17.39%.Benzyl[(3aR,5R,6S,6aR)-6-azido-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl]acetate 12
To a solution of 11 (0.210 g, 0.86 mmol) in CH3CN (5 mL) was added Et3N (0.27 mL, 1.93 mmol), CbzCl (0.245 mL, 1.72 mmol) and DMAP (0.05 g, 0.43 mmol). After stirring for 2 h at 25 °C, another portion of CbzCl (0.120 mL, 0.86 mmol) and DMAP (0.03 g, 0.22 mmol) were added and the mixture stirred overnight. It was concentrated in vacuo and extracted with EtOAc (3 × 15 mL). The combined organic extracts were sequentially washed with aqueous NaHCO3 (5 mL) and water (5 mL), and dried. Concentration of extract and column chromatography (silica gel, 5% EtOAc/hexane) of the residue afforded 12 (0.180 g, 63%) as a viscous liquid. Rf = 0.51 (10% EtOAc/hexane); [α]25D −28.5 (c 1.0, CHCl3); νmax/cm−1: 2110, 1734 cm−1; 1H NMR: δ 7.34 (s, 5H), 5.84 (d, J = 3.7 Hz, 1H), 5.14 (s, 2H), 4.65 (d, J = 3.7 Hz, 1H), 4.59 (ddd, J = 8.2, 6.3, 3.3 Hz, 1H), 4.09 (d, J = 3.3 Hz, 1H), 2.96–2.66 (m, 2H), 1.49 (s, 3H), 1.31 (s, 3H); 13C NMR: δ 170.0, 135.5, 128.6, 128.4, 128.2, 112.2, 104.2, 83.6, 75.5, 66.8, 66.7, 33.9, 26.5, 26.2; ESI-MS: calcd for [C16H19N3O5 + Na]+: 356.12 Da. Found: 355.89 Da. Anal. calcd for C16H19N3O5: C, 57.65; H, 5.75; N, 12.61%. Found: C, 57.63; H, 5.78; N, 12.70%.(2S,3R)-2-Azido-5-(benzyloxy)-3-hydroxy-5-oxopentanoic acid 13
As described earlier, 12 (0.100 g, 0.30 mmol) was deacetalized with TFA–H2O (3 mL, 3:2), the resultant diol cleaved with NaIO4 (0.072 g, 0.33 mmol) in 10% aqueous acetone (5 mL) followed by oxidation with NaH2PO4 (0.01 g, 0.06 mmol), 30% H2O2 (50 μL, 0.30 mmol) and NaClO2 (0.05 g, 0.49 mmol). The product was finally deformylated with aqueous saturated NaHCO3 (1 mL). Usual workup and column chromatography (silica gel, 2% MeOH/CHCl3) of the residue afforded 13 (0.060 g, 71% over four steps) as a thick liquid. Rf = 0.31 (10% MeOH/CHCl3); [α]25D −20.0 (c 1.01, CHCl3); νmax/cm−1: 2112, 1725 cm−1; 1H NMR: δ 7.33 (s, 5H), 6.91 (broad s, D2O exchangeable, 2H), 5.13 (s, 2H), 4.71–4.60 (m, 1H), 3.96 (d, J = 2.1 Hz, 1H), 2.80 (dd, J = 16.6, 8.3 Hz, 1H), 2.64 (dd, J = 16.6, 4.7 Hz, 1H); 13C NMR: δ 172.4, 171.7, 135.1, 128.6, 128.5, 128.3, 68.7, 67.0, 64.8, 38.0. Anal. calcd for C12H13N3O5: C, 51.61; H, 4.69; N, 15.05%. Found: C, 51.58; H, 4.72; N, 15.13%.
5-Benzyl 1-prop-2-en-1-yl(2S,3R)-2-azido-3-hydroxypentanedioate 14
Following the procedure used for 9, the acid 13 (0.078 g, 0.27 mmol) was subjected to allylation using allyl bromide (29.0 μL) and NaHCO3 (0.056 g) in DMF (1 mL). Usual workup and column chromatography (silica gel, 5% EtOAc/hexane) of the residue afforded 14 (0.075 g, 87%) as a thick liquid. Rf = 0.38 (20% EtOAc/hexane); [α]25D −24.3 (c 1.08, CHCl3); νmax/cm−1: 2108, 1742 cm−1; 1H NMR: δ 7.34 (s, 5H), 6.05–5.80 (m, 1H), 5.42–5.29 (m, 2H), 5.15 (s, 2H), 4.72 (broad d, J = 5.8 Hz, 2H), 4.65–4.53 (m, 1H), 3.87 (d, J = 3.1 Hz, 1H), 3.14 (s, D2O exchangeable, 1H), 2.79 (dd, J = 16.7, 8.4 Hz, 1H), 2.62 (dd, J = 16.7, 4.5 Hz, 1H); 13C NMR: δ 171.5, 168.2, 135.2, 131.0, 128.7, 128.5, 128.3, 119.4, 68.9, 66.9, 66.7, 64.9, 38.0; ESI-MS: calcd for [C15H17N3O5 + Na]+: 342.10 Da. Found: 341.85 Da. Anal. calcd for C15H17N3O5: C, 56.42; H, 5.37; N, 13.16%. Found: C, 56.48; H, 5.43; N, 13.23%.(2S,3R)-3-Hydroxy-L-glutamic acid hydrochloride 1a
A mixture of 13 (0.085 g, 0.31 mmol) and 10% Pd–C (0.02 g) in methanolic HCl (10 mL) was stirred for 12 h under H2 (80 psi). The catalyst was filtered through Celite-545 and washed with MeOH (3 × 10 mL), concentrated and the residue dried in vacuo to afford 1a (0.048 g, 95%) as a semisolid. [α]25D +14.7 (c 1.02, H2O); νmax/cm−1: 3577, 1737 cm−1; 1H NMR: δ 4.66–4.58 (m, 1H), 4.10 (d, J = 3.3 Hz, 1H), 2.87 (dd, J = 16.4, 4.0 Hz, 1H), 2.72 (dd, J = 16.4, 8.7 Hz, 1H); 13C NMR: δ 174.1, 170.3, 65.5, 57.3, 38.4; ESI-MS: calcd for [C5H9NO5 + Na]+: 186.04 Da. Found: 186.10 Da. Anal. calcd for C5H10ClNO5: C, 30.09; H, 5.05; N, 7.02%. Found: C, 30.13; H, 5.10; N, 7.13%.2-[(3aR,5R,6S,6aR)-6-Azido-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl]-N-benzylacetamide 15
To a solution of 11 (0.080 g, 0.33 mmol) in DMF at 25 °C was added HBTU (0.14 g, 0.37 mmol), HOBt monohydrate (0.06 g, 0.37 mmol) and DIEA (0.17 mL, 0.99 mmol). After stirring for 5 min, benzylamine (0.04 mL, 0.38 mmol) in DMF (0.40 mL) was added, and the mixture stirred for an additional 8 h. The mixture was concentrated in vacuo, the residue extracted with EtOAc (3 × 20 mL), the organic extract washed with water (3 × 5 mL) and brine (1 × 5 mL), and dried. Concentration of the extract in vacuo, and column chromatography (silica gel, 12% EtOAc/hexane) of the residue yielded 15 (0.068 g, 62%) as a thick liquid. Rf = 0.55 (50% EtOAc/hexane); [α]25D −24.0 (c 1.05, CHCl3); νmax/cm−1: 2103, 1689 cm−1; 1H NMR: δ 7.31–7.23 (m, 2H), 7.22–7.17 (m, 3H), 6.29 (broad s, D2O exchangeable, 1H), 5.79 (d, J = 3.7 Hz, 1H), 4.59 (d, J = 3.7 Hz, 1H), 4.56–4.49 (m, 1H), 4.41 (dd, J = 14.8, 5.8 Hz, 1H), 4.34 (dd, J = 14.8, 5.6 Hz, 1H), 3.98 (d, J = 3.0 Hz, 1H), 2.62 (dd, J = 15.2, 7.7 Hz, 1H), 2.53 (dd, J = 15.2, 5.8 Hz, 1H), 1.43 (s, 3H), 1.25 (s, 3H); 13C NMR: δ 170.0, 137.7, 128.7, 127.6, 127.5, 112.4, 104.3, 83.4, 76.0, 67.1, 43.7, 36.2, 26.5, 26.2; ESI-MS: calcd for [C16H20N4O4 + Na]+: 355.13 Da. Found: 354.93 Da. Anal. calcd for C16H20N4O4: C, 57.82; H, 6.07; N, 16.86%. Found: C, 57.80; H, 6.05; N, 16.93%.(2S,3R)-2-Azido-5-(benzylamino)-3-hydroxy-5-oxopentanoic acid 16
Following the procedure used for the synthesis of 13, compound 15 (0.110 g, 0.33 mmol) was deacetalized with TFA–H2O (3 mL, 3:2), the resultant diol cleaved with NaIO4 (0.08 g, 0.37 mmol) in 10% aqueous acetone (5 mL) followed by oxidation with NaH2PO4 (0.01 g), 30% H2O2 (35 μL) and NaClO2 (0.05 g). The product was finally deformylated with aqueous saturated NaHCO3 (1 mL). Usual workup and column chromatography (silica gel, 10% MeOH/CHCl3) of the residue afforded 16 (0.067 g, 72% in four steps) as a thick liquid. Rf = 0.35 (30% MeOH/CHCl3); [α]25D −19.0 (c 1.04, CHCl3); νmax/cm−1: 3570, 2107, 1737, 1685 cm−1; 1H NMR: δ 7.34–7.23 (m, 5H), 7.18 (broad s, D2O exchangeable, 1H), 4.43 (s, 1H), 4.34 (s, 2H), 3.74 (s, 1H), 2.49 (s, 2H); 13C NMR: δ 173.9, 172.0, 138.4, 128.1, 127.1, 126.7, 69.4, 67.5, 42.7, 39.9. Anal. calcd for C12H14N4O4: C, 51.80; H, 5.07; N, 20.13%. Found: C, 51.87; H, 5.13; N, 20.25%.
Prop-2-en-1-yl(2S,3R)-2-azido-5-(benzylamino)-3-hydroxy-5-oxopentanoate 17
Following the procedure described earlier, 16 (0.06 g, 0.22 mmol) was reacted with allyl bromide (20 μL) in the presence of NaHCO3 (0.04 g) in DMF (2 mL). Usual workup and column chromatography (silica gel, 8% EtOAc/hexane) of the residue gave 17 (0.055 g, 79%) as a thick liquid. Rf = 0.41 (30% EtOAc/hexane); [α]25D −16.4 (c 0.55, CHCl3); νmax/cm−1: 2105, 1744 cm−1; 1H NMR: δ 7.38–7.17 (m, 5H), 6.13–5.69 (m, partially D2O exchangeable, 2H), 5.44–5.15 (m, 2H), 4.67 (broad d, J = 5.8 Hz, 2H), 4.62–4.46 (m, 1H), 4.38 (d, J = 5.7 Hz, 2H), 3.75 (d, J = 3.2 Hz, 1H), 2.58 (dd, J = 15.4, 8.9 Hz, 1H), 2.37 (dd, J = 15.4, 3.8 Hz, 1H), 1.58 (broad s, D2O exchangeable, 1H); 13C NMR: δ 170.8, 168.4, 137.6, 131.1, 128.8, 127.8, 127.7, 119.4, 69.6, 66.7, 65.1, 43.7, 39.1; ESI-MS: calcd for [C15H18N4O4 + Na]+: 341.12 Da. Found: 340.92 Da. Anal. calcd for C15H18N4O4: C, 56.60; H, 5.70; N, 17.60%. Found: C, 56.55; H, 5.68; N, 17.69%.(1S,2R)-4-(Benzylamino)-1-carboxy-2-hydroxy-4-oxobutan-1-aminium chloride 18
Catalytic hydrogenation of 16 (0.048 g, 0.17 mmol) over 10% Pd–C (0.015 g) in MeOH (5 mL) using H2 (80 psi) gave 18 (0.044 g, 89%) as a semi-solid. [α]25D +22.0 (c 1.00, CHCl3); νmax/cm−1: 1713, 1406 cm−1; 1H NMR: δ 7.44 (d, J = 3.9 Hz, 2H), 7.38 (s, 3H), 4.72–4.64 (m, 1H), 4.44 (s, 2H), 4.19 (d, J = 3.7 Hz, 1H), 2.82–2.76 (m, 1H), 2.70 (dd, J = 14.8, 8.8 Hz, 1H); 13C NMR: δ 173.6, 171.7, 139.5, 130.5, 129.2, 129.0, 67.8, 59.0, 44.8, 41.8; ESI-MS: calcd for [C12H16N2O4 + H]+: 253.11 Da. Obsd: 253.90 Da. Anal. calcd for C12H17ClN2O4: C, 49.92; H, 5.93; N, 9.70%. Found: C, 49.88; H, 5.96; N, 9.78%.2-[(3aR,5R,6S,6aR)-6-Azido-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl]acetamide 19
To a solution of 11 (0.210 g, 0.87 mmol) in CH3CN (10 mL) was added (Boc)2O (0.246 g, 1.13 mmol), and NH4HCO3 (0.161 g, 2.04 mmol) to give a cloudy mixture. After adding pyridine (0.05 mL, 0.60 mmol), the mixture was stirred at room temperature till completion of the reaction (∼for 5 h, cf. TLC). The mixture was concentrated in vacuo, the residue extracted with EtOAc (3 × 15 mL), the organic extract washed with water (5 mL) and dried. Concentration of the extract in vacuo, and column chromatography (silica gel, 30% EtOAc/hexane) of the residue afforded 19 (0.189 g, 90%) as colorless crystals. mp: 122–125 °C; Rf = 0.45 (80% EtOAc/hexane); [α]25D −23.1 (c 1.18, MeOH); νmax/cm−1: 2111, 1665 cm−1; 1H NMR: δ 5.81 (d, J = 3.7 Hz, 1H), 4.72 (d, J = 3.7 Hz, 1H), 4.56 (td, J = 6.9, 3.1 Hz, 1H), 4.04 (d, J = 3.1 Hz, 1H), 2.63 (dd, J = 15.2, 7.3 Hz, 1H), 2.55 (dd, J = 15.2, 6.6 Hz, 1H), 1.45 (s, 3H), 1.29 (s, 3H); 13C NMR: δ 172.1, 110.2, 102.7, 81.9, 74.6, 65.6, 33.0, 23.8, 23.4; ESI-MS: calcd for [C9H14N4O4 + Na]+: 265.09 Da. Found: 265.90 Da. Anal. calcd for C9H14N4O4: C, 44.63; H, 5.83; N, 23.13%. Found: C, 44.59; H, 5.87; N, 23.25%.(2S,3R)-5-Amino-2-azido-3-hydroxy-5-oxopentanoic acid 20
Following the procedure used for the synthesis of 16, compound 19 (0.08, 0.33 mmol) was deacetalized with TFA–H2O (3 mL, 3:2), the resultant diol subjected to oxidative cleavage with NaIO4 (0.08 g, 0.37 mmol) followed by oxidation with NaH2PO4 (0.01 g), 30% H2O2 (35 μL) and NaClO2 (0.05 g). The product was deformylated using NaHCO3 (1 mL) in THF (5 mL). Usual workup and purification by column chromatography (silica gel, 10% MeOH/CHCl3) of the residue gave 20 (0.04 g, 66% over four steps) as a thick liquid. Rf = 0.45 (30% MeOH/CHCl3); [α]25D −14.4 (c 1.00, MeOH); νmax/cm−1: 2110, 1690 cm−1; 1H NMR: δ 4.50 (broad s, 1H), 3.85 (d, J = 1.5 Hz, 1H), 2.49 (dd, J = 14.7, 8.2 Hz, 1H), 2.42 (dd, J = 14.7, 5.0 Hz, 1H); 13C NMR: δ 174.5, 170.6, 69.0, 65.3, 39.4. Anal. calcd for C5H8N4O4: C, 31.92; H, 4.29; N, 29.78%. Found: C, 31.95; H, 4.26; N, 29.87%.
(2S,3R)-3-Hydroxy-L-glutamine hydrochloride 1b
The procedure for transforming 13 to 1a was followed for the catalytic hydrogenation of 20 (0.02, 0.11 mmol) using 10% Pd/C (0.015 g) and H2 (80 psi) to give 1b as a semisolid (0.019 g, 90%). [α]25D +9.1 (c 1.08, H2O); νmax/cm−1: 3440, 1708 cm−1; 1H NMR: δ 4.71–4.60 (m, 1H), 4.20 (d, J = 3.8 Hz, 1H), 2.77 (dd, J = 15.1, 4.1 Hz, 1H), 2.67 (dd, J = 15.1, 8.8 Hz, 1H); 13C NMR: δ 174.8, 169.9, 65.8, 57.1, 39.3; ESI-MS: calcd for [C5H10N2O4 + Na]+: 185.05 Da. Found: 185.00 Da. Anal. calcd for C5H11ClN2O4: C, 30.24; H, 5.58; N, 14.11%. Found: C, 30.29; H, 5.64; N, 14.23%.(3aR,4aR,7aS,7bR)-2,2-Dimethylhexahydro-6H-[1,3]dioxolo[4,5]furo[3,2-b]pyrrol-6-one 21
A mixture of 6 (0.300 g, 1.16 mmol), 10% Pd/C (0.03 g) and HCO2NH4 (0.220 g, 3.50 mmol) in MeOH (15 mL) was refluxed for 2 h till completion of the reaction (cf. TLC). The mixture was filtered through Celite-545, the residue washed with MeOH (10 mL) and concentrated in vacuo. Column chromatography (silica gel, EtOAc) of the residue yielded 21 (0.210 g, 91%) as a white solid. mp: 174–176 °C; Rf = 0.30 (EtOAc); [α]25D +24.8 (c 1.00, CHCl3); νmax/cm−1: 1658, 1413 cm−1; 1H NMR: δ 7.45 (broad s, D2O exchangeable, 1H), 5.88 (d, J = 3.8 Hz, 1H), 4.90 (t, J = 3.8 Hz, 1H), 4.58 (d, J = 3.8 Hz, 1H), 4.08 (d, J = 4.2 Hz, 1H), 2.60–2.32 (m, 2H), 1.46 (s, 3H), 1.27 (s, 3H); 13C NMR: δ 177.0, 112.1, 106.0, 83.2, 77.9, 63.9, 38.0, 26.9, 26.4. Anal. calcd for C9H13NO4: C, 54.26; H, 6.58; N, 7.03%. Found: C, 54.29; H, 6.63; N, 7.10%.(3aR,4aR,7aS,7bR)-2,2-Dimethylhexahydro-3aH-[1,3]dioxolo[4,5]furo[3,2-b]-N-carboxybenzylpyrrole 22
To a stirred and ice-cold suspension of LiAlH4 (0.071 g, 1.88 mmol) in dry THF (5 mL) was added 21 (0.150 g, 0.75 mmol) in dry THF (10 mL) in 5 min. The mixture was stirred further for 15 min at 0 °C, allowed to attain room temperature (25 °C), and then refluxed. After 3 h, the mixture was cooled to 25 °C, EtOAc (7 mL) added into it slowly followed by aqueous saturated NH4Cl (1 mL) and stirred for 1 h. It was filtered through Celite-545 by washing with 20% MeOH–EtOAc, and the filtrate evaporated in vacuo to give the corresponding amine. The amine was dissolved in MeOH–water (3:1, 10 mL), cooled to 0 °C, NaHCO3 (0.176 g, 2.10 mmol) and CbzCl (0.25 mL, 1.75 mmol) were successively added into it. After 3 h, the mixture was concentrated in vacuo, and the residue extracted with CH2Cl2 (3 × 10 mL). The organic extract was dried, concentrated in vacuo, and the residue purified by column chromatography (silica gel, 7% EtOAc/hexane) to give 22 (0.136 g, 57% over two steps) as a colorless thick liquid. Rf = 0.50 (20% EtOAc/hexane); [α]25D −55.6 (c 1.06, CHCl3); νmax/cm−1: 1671, 1420 cm−1; 1H NMR: δ 7.49–7.25 (m, 5H), 5.80 (s, 1H), 5.20–5.01 (m, 2H), 4.92–4.60 (m, 2H), 4.20 (s, 1H), 3.77–3.60 (m, 1H), 3.40–3.23 (m, 1H), 2.06 (dd, J = 13.6, 6.0 Hz, 1H), 1.91–1.69 (m, 1H), 1.49 (s, 3H), 1.28 (s, 3H); 13C NMR: δ 154.4, 136.6, 128.5, 128.0, 127.9, 111.8, 106.0, 84.5, 82.2, 67.6, 67.0, 45.5, 30.0, 27.1, 26.5; ESI-MS: calcd for [C17H21NO5 + Na]+: 342.13 Da. Found: 341.93 Da. Anal. calcd for C17H21NO5: C, 63.94; H, 6.63; N, 4.39%. Found: C, 63.89; H, 6.67; N, 4.47%.
(2S,3R)-3-Hydroxyproline hydrochloride 1c
Following the procedure used for the synthesis of 16, compound 22 (0.160 g, 0.50 mmol) was deacetalized with TFA–H2O (3 mL, 3:2), the resultant diol subjected to oxidative cleavage with NaIO4 (0.110 g, 0.50 mmol), followed by oxidation with NaH2PO4 (0.01 g), 30% H2O2 (35 μL), NaClO2 (0.05 g) and subsequent deformylation with aqueous saturated NaHCO3 (1 mL) in THF (5 mL). The resultant acid was hydrogenated with H2 (80 psi) over 10% Pd–C (0.015 g) to afford 1c (0.045 g, 54% over five steps) as a pale-yellow semi-solid. [α]25D −12.3 (c 1.04, H2O); νmax/cm−1: 3576, 1715 cm−1; 1H NMR: δ 4.86 (t, J = 4.0 Hz, 1H), 4.44 (d, J = 4.0 Hz, 1H), 3.50–3.71 (m, 2H), 2.39–2.29 (m, 1H), 2.26–2.18 (m, 1H); 13C NMR: δ 169.2, 70.8, 66.3, 44.0, 32.9. Anal. calcd for C5H10ClNO3: C, 35.83; H, 6.01; N, 8.36%. Found: C, 35.80; H, 6.07; N, 8.44%.
Prop-2-en-1-yl(2S,3R)-N-carboxybenzyl-3-hydroxypyrrolidine-2-carboxylate 23
As described before, 22 (0.064 g, 0.20 mmol) was deacetalized using TFA–H2O (3 mL, 3:2), the resultant diol oxidatively cleaved with NaIO4 (0.047 g, 0.22 mmol) followed by oxidation with NaH2PO4 (0.006 g), 30% H2O2 (21 μL), NaClO2 (0.03 g), and the formyl group unmasked with aqueous NaHCO3 (1 mL) in THF (5 mL). The resultant crude acid was dried in vacuo and allylated with allyl bromide (21.6 μL) and NaHCO3 (0.04 g) in DMF (3 mL). The mixture was concentrated in vacuo, the residue extracted with EtOAc (3 × 10 mL), the organic extract washed with water (2 × 5 mL) and dried. Concentration of the extract in vacuo, and column chromatography of the residue (silica gel, 15% EtOAc/hexane) gave 23 (0.040 g, 65% over five steps) as a thick liquid. Rf = 0.36 (50% EtOAc/n-hexane); [α]25D +28.00 (c 1.00, CHCl3); νmax/cm−1: 1715, 1215 cm−1; 1H NMR: δ 7.45–7.22 (m, 5H), 6.08–5.60 (m, 1H), 5.48–4.91 (m, 4H), 4.86–4.34 (m, 4H), 3.81–3.40 (m, 2H), 2.84 (broad s, D2O exchangeable, 1H), 2.24–1.82 (m, 2H); 13C NMR: δ 169.8, 154.4, 136.2, 131.8, 128.4, 128.0, 127.8, 118.5, 72.2, 67.2, 65.9, 63.8, 44.4, 32.0; ESI-MS: calcd for [C16H19NO5 + Na]+: 328.11 Da. Found: 327.96 Da. Anal. calcd for C16H19NO5: C, 62.94; H, 6.27; N, 4.59%. Found: C, 63.01; H, 6.25; N, 4.66%.
(2R,3R)-2-(Hydroxymethyl)-N-carboxybenzylpyrrolidin-3-ol 24
A solution of 22 (0.100 g, 0.31 mmol) in TFA–H2O (3.00 mL, 3:2) was stirred at to 0 to 10 °C for 1.5 h. TFA was removed azeotropically with toluene in vacuo to afford the hemiacetal as a thick liquid. To a cooled (0 °C) solution of the crude hemiacetal in acetone–water (9:1, 5 mL) was added NaIO4 (0.073 g, 0.34 mmol). After stirring for 30 min, the reaction mixture was concentrated in vacuo, and the residue extracted with CHCl3 (3 × 10 mL) to get the crude aldehyde (0.09 g) as a thick liquid. This was dissolved in THF–H2O (4:1, 5 mL), cooled to 5 °C and NaBH4 (0.015 g, 0.40 mmol) in H2O (0.5 mL) was added to it. After stirring for 30 min, the mixture was concentrated in vacuo, the residue extracted with EtOAc (2 × 10 mL), the organic extract washed with water (2 × 5 mL) and dried. Concentration of the extract in vacuo, and column chromatography of the residue (silica gel, 20% EtOAc/hexane) yielded 24 (0.05 g, 63% over three steps) as a thick liquid. Rf = 0.30 (60% EtOAc/hexane); [α]25D −11.5 (c 1.20, CHCl3); νmax/cm−1: 1673, 1421 cm−1; 1H NMR: δ 7.41–7.28 (m, 5H), 5.12 (q, J = 12.4 Hz, 2H), 4.51 (d, J = 3.7 Hz, 1H), 4.09–3.80 (m, 3H), 3.54 (t, J = 6.3 Hz, 2H), 2.85–2.34 (m, D2O exchangeable, 2H), 2.11–1.85 (m, 2H); 13C NMR: δ 154.5, 136.4, 128.5, 128.1, 128.0, 72.8, 67.2, 61.9, 44.6, 32.9; ESI-MS: calcd for [C13H17NO4 + Na]+: 274.10 Da. Found: 274.11 Da. Anal. calcd for C13H17NO4: C, 62.14; H, 6.82; N, 5.57%. Found: C, 62.18; H, 6.87; N, 5.66%.
(2R,3R)-2-(Hydroxymethyl)pyrrolidin-3-ol 2
A mixture of 24 (0.080 g, 0.31 mmol) and 10% Pd/C (0.02 g) in MeOH (10 mL) was stirred under H2 (80 psi) for 6 h. The catalyst was filtered through Celite-545 by washing with MeOH (20 mL), and the filtrate concentrated in vacuo to afford 2 (0.030 g, 84%) as a thick liquid. Rf = 0.15 (40% MeOH/CHCl3); [α]25D +11.5 (c 1.14, H2O); νmax/cm−1: 3480 cm−1; 1H NMR: δ 4.57 (s, 1H), 3.99 (dd, J = 12.0, 5.1 Hz, 1H), 3.87 (dd, J = 12.0, 8.2 Hz, 1H), 3.58–3.51 (m, 1H), 3.50–3.40 (m, 1H), 3.38–3.32 (m, 1H), 2.30–2.20 (m, 1H), 2.11–2.03 (m, 1H); 13C NMR: δ 70.5, 64.5, 58.6, 43.1, 33.1; ESI-MS: calcd for [C5H11NO2 + H]: 118.08 Da. Found: 118.19 Da. Anal. calcd for C5H11NO2: C, 51.26; H, 9.46; N, 11.96%. Found: C, 51.33; H, 9.38; N, 12.05%.References
- (a) Amino acids, Peptides, and Proteins, ed. R. C. Sheppard, The Chemical Society, London, 1972–1981 Search PubMed; (b) Amino acids, Peptides, and Proteins, ed. G. T. Young, The Chemical Society, London, 1968–1971 Search PubMed; (c) R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS PubMed; (d) A. Varki, Glycobiology, 1993, 3, 97–130 CrossRef CAS PubMed; (e) C.-H. Wong, Acc. Chem. Res., 1999, 32, 376–385 CrossRef CAS; (f) C. T. Walsh, Antibiotics: Actions, Origins, Resistance, ASM Press, Washington, DC, 2003 Search PubMed; (g) C. T. Walsh, Post Translational Modifications of Proteins: Expanding Natures Inventory, Roberts and Company, Englewood, 2006 Search PubMed.
- (a) P. E. Dawson and S. B. H. Kent, Annu. Rev. Biochem., 2000, 69, 923–960 CrossRef CAS PubMed; (b) S. B. H. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC.
- (a) J. K. Thottathil, J. L. Moniot, R. H. Mueller, M. K. Y. Wong and T. P. Kissick, J. Org. Chem., 1986, 51, 3140–3143 CrossRef CAS; (b) J. K. Thottathil and J. L. Moniot, Tetrahedron Lett., 1986, 27, 151–154 CrossRef CAS; (c) J. Krapcho, C. Turk, D. W. Cushman, J. R. Powell, J. M. DeForrest, E. R. Spitzmiller, D. S. Karanewsky, M. Duggan, G. Rovnyak, J. Schwartz, S. Natarajan, J. D. Godfrey, D. E. Ryono, R. Neubeck, K. S. Atwal and E. W. Petrillo Jr, J. Med. Chem., 1988, 31, 1148–1160 CrossRef CAS PubMed; (d) A. M. P. Koskinen and H. Rapoport, J. Org. Chem., 1989, 54, 1859–1866 CrossRef CAS; (e) P. Remuzon, Tetrahedron, 1996, 52, 13803–13835 CrossRef CAS; (f) K. Hashimoto, Y. Shima and H. Shirahama, Heterocycles, 1996, 42, 489–492 CrossRef CAS; (g) Q. Wang, N. A. Sasaki and P. Potier, Tetrahedron, 1998, 54, 15759–15780 CrossRef CAS; (h) T. Goodman and L. Moroder, in Synthesis of Peptides and Peptidomimetics, Houben-Weyl, Thieme, Stuttgart, Germany, 2003, vol. E22c, pp. 215–271 Search PubMed.
- (a) M. R. Levengood, P. J. Knerr, T. J. Oman and W. A. van der Donk, J. Am. Chem. Soc., 2009, 131, 12024–12025 CrossRef CAS PubMed; (b) P. J. Knerr and W. A. van der Donk, J. Am. Chem. Soc., 2012, 134, 7648–7651 CrossRef CAS PubMed; (c) J. Vagner, H. Qu and V. J. Hruby, Curr. Opin. Chem. Biol., 2008, 12, 292–296 CrossRef CAS PubMed.
- (a) P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–778 CAS; (b) L. Z. Yan and P. E. Dawson, J. Am. Chem. Soc., 2001, 123, 526–533 CrossRef CAS PubMed; (c) N. A. McGrath and R. T. Raines, Chem. Res., 2011, 44, 752–761 CrossRef CAS PubMed.
- (a) R. J. Payne and C.-H. Wong, Chem. Commun., 2010, 46, 21–43 RSC; (b) P. Siman and A. Brik, Org. Biomol. Chem., 2012, 10, 5684–5697 RSC; (c) L. Raibaut, N. Ollivier and O. Melnyk, Chem. Soc. Rev., 2012, 41, 7001–7015 RSC; (d) N. Metanis, E. Keinan and P. E. Dawson, Angew. Chem., Int. Ed., 2010, 49, 7049–7052 CrossRef CAS PubMed; (e) S. D. Townsend, Z. Tan, S. Dong, S. Shang, J. A. Brailsford and S. J. Danishefsky, J. Am. Chem. Soc., 2012, 134, 3912–3916 CrossRef CAS PubMed.
- (a) D. Crich and A. Banerjee, J. Am. Chem. Soc., 2007, 129, 10064–100065 CrossRef CAS PubMed; (b) C. Haase, H. Rohde and O. Seitz, Angew. Chem., Int. Ed., 2008, 47, 6807–6810 CrossRef CAS PubMed; (c) J. Chen, Q. Wan, Y. Yuan, J. Zhu and S. J. Danishefsky, Angew. Chem., Int. Ed., 2008, 47, 8521–8524 CrossRef CAS PubMed.
- (a) R. Yang, K. K. Pasunooti, F. Li, X.-W. Liu and C.-F. Liu, J. Am. Chem. Soc., 2009, 131, 13592–13593 CrossRef CAS PubMed; (b) J. Chen, P. Wang, J. Zhu, Q. Wan and S. J. Danishefsky, Tetrahedron, 2010, 66, 2277–2283 CrossRef CAS PubMed; (c) J. Chen, P. Wang, J. Zhu, Q. Wan and S. J. Danishefsky, Tetrahedron, 2010, 66, 2277–2283 CrossRef CAS PubMed; (d) Z. Tan, S. Shang and S. J. Danishefsky, Angew. Chem., Int. Ed., 2010, 49, 9500–9503 CrossRef CAS PubMed; (e) S. Shang, Z. Tan, S. Dong and S. J. Danishefsky, J. Am. Chem. Soc., 2011, 133, 10784–10786 CrossRef CAS PubMed; (f) P. Siman, S. V. Karthikeyan and A. Brik, Org. Lett., 2012, 14, 1520–1523 CrossRef CAS PubMed; (g) L. R. Malins, K. M. Cergol and R. J. Payne, ChemBioChem, 2013, 14, 559–563 CrossRef CAS PubMed; (h) R. E. Thompson, B. Chan, L. Radom, K. A. Jolliffe and R. J. Payne, Angew. Chem., Int. Ed., 2013, 52, 9723–9727 CrossRef CAS PubMed; (i) K. M. Cergol, R. E. Thompson, L. R. Malins, P. Turner and R. J. Payne, Org. Lett., 2014, 16, 290–293 CrossRef CAS PubMed; (j) S. D. Townsend, Z. Tan, S. Dong, S. Shang, J. A. Brailsford and S. J. Danishefsky, J. Am. Chem. Soc., 2012, 134, 3912–3916 CrossRef CAS PubMed.
- (a) D. D. Van Slyke and A. Hiller, Proc. Natl. Acad. Sci. U. S. A., 1921, 7, 185–186 CrossRef CAS PubMed; (b) J. C. Sheeham, K. Maeda, A. K. Sen and J. A. Stock, J. Am. Chem. Soc., 1962, 84, 1303–1305 CrossRef; (c) W. Traub and K. A. Piez, Adv. Protein Chem., 1971, 25, 243–352 CrossRef CAS PubMed; (d) H. Maehr, C.-M. Liu, N. J. Palleroni, J. Smallheer, L. Todan, T. H. Williams and J. F. Blount, J. Antibiot., 1986, 39, 17–25 CrossRef CAS PubMed; (e) T. A. Smitka, J. B. Deeter, A. H. Hunt, F. P. Mertz, R. M. Ellis, L. D. Boeck and R. C. Yao, J. Antibiot., 1988, 41, 726–733 CrossRef CAS PubMed; (f) A. A. Tymiak, T. J. McCormick and S. E. Unger, J. Org. Chem., 1989, 54, 1149–1157 CrossRef CAS; (g) S. Omura, T. Fujimoto, K. Otoguro, R. Koriguchi, H. Tanaka and Y. Sasaki, J. Antibiot., 1991, 44, 113–116 CrossRef CAS PubMed; (h) S. Omuro, K. Matsuzaki, T. Fujimoto, K. Kosuge, T. Furuya, S. Fujita and A. Nakagawa, J. Antibiot., 1991, 44, 117–118 CrossRef; (i) S. Chatterjee, D. K. Chatterjee, R. H. Jani, J. Blumbach, B. N. Ganguli, N. Klesel, M. Limbert and G. Siebert, J. Antibiot., 1992, 45, 839–845 CrossRef CAS PubMed; (j) For amino acids as enzyme inhibitors see: C. Walsh, Tetrahedron, 1982, 38, 871–909 CrossRef CAS; (k) A. I. Ayi and R. Guedj, J. Chem. Soc., Perkin Trans. 1, 1983, 2045–2051 RSC.
- For mono target oriented hydroxy amino acid synthesis see: For-leu: (a) D. Seebach, E. Juaristi, D. D. Miller, C. Schickli and T. Weber, Helv. Chim. Acta, 1987, 70, 237–261 CrossRef CAS; (b) D. A. Evans, E. B. Sjogren, A. E. Weber and R. E. Conn, Tetrahedron Lett., 1987, 28, 39–42 CrossRef CAS; (c) M. E. Jung and Y. H. Jung, Tetrahedron Lett., 1989, 48, 6637–6640 CrossRef; (d) C. G. Cald Well and S. S. Bondy, Synthesis, 1990, 34–35 CrossRef CAS; (e) E. J. Corey, D. H. Lee and S. Choi, Tetrahedron Lett., 1992, 33, 6735–6738 CrossRef CAS; (f) J. S. Yadav, S. Chandrasekhar, R. Y. Reddy and A. V. R. Rao, Tetrahedron, 1995, 51, 2749–2754 CrossRef CAS ; For Pro:; (g) S. Hanessian and S. P. Sahoo, Can. J. Chem., 1984, 62, 1400–1402 CrossRef CAS; (h) J. Cooper, P. T. Gallagher and D. W. Knight, J. Chem. Soc., Chem. Commun., 1988, 509–510 RSC; (i) N. B. Kalamkar, V. M. Kasture and D. D. Dhavale, Tetrahedron Lett., 2010, 51, 6745–6747 CrossRef CAS and references cited therein. For a review on 5-hydroxy-lysine see:; (j) K. R. Herbert, G. M. Williams, G. J. S. Cooper and M. A. Brimble, Org. Biomol. Chem., 2012, 10, 1137–1144 RSC ; For Lys:; (k) P. F. Hughes, S. H. Smith and J. T. Olson, J. Org. Chem., 1994, 59, 5799–5802 CrossRef CAS ; For Glu:; (l) T. Kunieda, T. Ishizuka, T. Higuchi and M. Hirobe, J. Org. Chem., 1988, 53, 3381–3383 CrossRef CAS; (m) H. Takahata, Y. Banba, M. Tajima and T. Momose, J. Org. Chem., 1991, 56, 240–245 CrossRef CAS; (n) H. Takahata, Y. Banba, M. Tajima and T. Momose, J. Org. Chem., 1991, 56, 240–245 CrossRef CAS; (o) S. Shiokawa, T. Ohta and S. Nozoe, Chem. Pharm. Bull., 1992, 40, 1398–1399 CrossRef; (p) N. D-Uomo, M. C. D-Giovannia, D. Misiti, G. Zappia and G. D. Monache, Liebigs Ann. Chem., 1994, 641–644 CrossRef; (q) L. Tamborini, P. Conti, A. Pinto, S. Colleoni, M. Gobbi and C. D. Micheli, Tetrahedron, 2009, 65, 6083–6089 CrossRef CAS.
- For multiple target oriented hydroxy amino acid synthesis see: (a) Y. N. Belokon, A. G. Bulychev, S. V. Vitt, Y. T. Struchkov, A. S. Batsanov, T. V. Timofeeva, V. A. Tsyryapkin, M. G. Ryzhov, L. A. Lysova, V. I. Bakhrnutov and V. M. Belikov, J. Am. Chem. Soc., 1985, 107, 4252–4259 CrossRef CAS; (b) S. S. Norio, B. M. Inabaf, T. Moriwaket and S. Torii, Tetrahedron Lett., 1985, 26, 5309–5312 CrossRef; (c) D. A. Evans, E. B. Sjogren, A. E. Weberm and R. E. Conn, Tetrahedron Lett., 1987, 28, 39–42 CrossRef CAS; (d) D. A. Evans and A. E. Weber, J. Am. Chem. Soc., 1987, 109, 7151–7157 CrossRef CAS; (e) M. Hirama, H. Hioki and S. Itô, Tetrahedron Lett., 1988, 29, 3125–3128 CrossRef CAS; (f) R. C. Roemmele and H. Rapoport, J. Org. Chem., 1989, 54, 1866–1875 CrossRef CAS; (g) M. A. Blaskovich and G. A. Lajoie, J. Am. Chem. Soc., 1993, 115, 5021–5030 CrossRef CAS; (h) M. A. Blaskovich and G. A. Lajoie, J. Am. Chem. Soc., 1993, 115, 5021–5030 CrossRef CAS; (i) Y. N. Belokon, K. A. Kochetkov, N. S. Ikonnikov, T. V. Strelkova, S. R. Harutyunyan and A. S. Saghiyan, Tetrahedron: Asymmetry, 2001, 12, 481–485 CrossRef CAS; (j) B. Ma, J. L. Parkinson and S. L. Castle, Tetrahedron Lett., 2007, 48, 2083–2086 CrossRef CAS PubMed; (k) S. G. Davies, A. M. Fletcher, A. B. Frost, J. A. Lee, P. M. Roberts and J. E. Thomson, Tetrahedron, 2013, 69, 8885–8898 CrossRef CAS; (l) S. C. Deshmukh and P. Talukdar, J. Org. Chem., 2014, 79, 11215–11225 CrossRef CAS PubMed; (m) Y. Singjunla, J. Baudoux and J. Rouden, Org. Lett., 2013, 15, 5770–5773 CrossRef CAS PubMed; (n) K. Makino, T. Goto, Y. Hiroki and Y. Hamada, Angew. Chem., Int. Ed., 2004, 43, 882–882 CrossRef CAS PubMed.
- (a) D. R. Spring, Org. Biomol. Chem., 2003, 1, 3867–3870 RSC; (b) D. S. Tan, Nat. Chem. Biol., 2005, 1, 74–84 CrossRef CAS PubMed.
- (a) K. S. A. Kumar, M. Haj-Yahya, D. Olschewski, H. A. Lashuel and A. Brik, Angew. Chem., Int. Ed., 2009, 48, 8090–8094 CrossRef PubMed; (b) Z. Harpaz, P. Siman, K. S. A. Kumar and A. Brik, ChemBioChem, 2010, 11, 1232–1235 CrossRef CAS PubMed; (c) K. S. A. Kumar, Bioorg. Med. Chem., 2013, 21, 3609–3613 CrossRef CAS PubMed; (d) V. P. Vyavahare, C. Chakraborty, B. Maity, S. Chattopadhyay, V. G. Puranik and D. D. Dhavale, J. Med. Chem., 2007, 50, 5519–5523 CrossRef CAS PubMed; (e) K. S. A. Kumar, J. S. Rathee, M. Subramanian and S. Chattopadhyay, J. Org. Chem., 2013, 78, 7406–7413 CrossRef CAS PubMed.
- N. Nepomniaschiy, V. Grimminger, A. Cohen, S. DiGiovanni, H. Lashuel and A. Brik, Org. Lett., 2008, 11, 5243–5524 CrossRef PubMed and references cited therein.
- (a) P. M. Headley and S. Grillner, Trends Pharmacol. Sci., 1990, 11, 205–211 CrossRef CAS PubMed; (b) H. Bräuner-Osborne, J. Egebjerg, E. Ø. Nielsen, U. Madsen and P. Krogsgaard-Larsen, J. Med. Chem., 2000, 43, 2609–2645 CrossRef; (c) J. N. C. Kew and J. A. Kemp, Psychopharmacology, 2005, 179, 4–29 CrossRef CAS PubMed.
- J. M. J. Tronchet, B. Gentile, J. Ojha-Poncet, G. Moret, D. Schwarzanbach and F. Barblat-Ray, Carbohydr. Res., 1977, 59, 87–93 CrossRef CAS.
- E. Dalcanale and F. Montanari, J. Org. Chem., 1986, 51, 567–569 CrossRef CAS.
- (a) V. Lee and M. S. Newman, Org. Synth., 1970, 50, 77 CrossRef CAS; (b) J. Mulzer, in Comprehensive Organic Functional Group Transformations, ed. A. R. Katritzky, O. Meth-Cohn and C. W. Rees, Pergamon Press, Oxford, 1995, vol. 5, pp. 146 and 276 Search PubMed; (c) J. Podlech and D. Seebach, Angew. Chem., Int. Ed., 1995, 34, 471–472 CrossRef CAS; (d) J. N. Tilekar, N. T. Patil and D. D. Dhavale, Synthesis, 2000, 3, 395–398 CrossRef; (e) C. W. Jefford, Q. Tang and A. Zaslona, J. Am. Chem. Soc., 1991, 113, 3513–3518 CrossRef CAS.
- (a) J. H. Lee, J. E. Kang, M. S. Yang, K. Y. Kang and K. H. Park, Tetrahedron, 2001, 57, 10071–10076 CrossRef CAS; (b) L. M. Mascavage, Q. Lu, J. Vey, D. R. Dalton and P. J. Carrol, J. Org. Chem., 2001, 66, 3621–3626 CrossRef CAS PubMed; (c) E. M. Dangerfield, C. H. Plunkett, B. L. Stocker and M. S. M. Timmer, Molecules, 2009, 14, 5298–5307 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra01340b |
| This journal is © The Royal Society of Chemistry 2015 |