
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poly(ether–amide) vs. poly(ether–imide) copolymers for post-combustion membrane separation processes
Alberto Tena*,
Sergey Shishatskiy and
Volkan Filiz
Helmholtz-Zentrum Geesthacht, Institute of Polymer Research, Max-Planck-Str.1, 21502 Geesthacht, Germany. E-mail: alberto.tena@hzg.de; Fax: +49 4152 872499; Tel: +49 4152 872448
First published on 19th February 2015
Abstract
This work is focused on the comparison between the commercial polyamide PEBAX® MH 1657 and a new set of synthetized polyimides with different polyethylene glycol lengths. The samples were synthesized with the same poly(ethylene oxide) (PEO) content (57 wt%) for comparison with the commercial polymer. All polymers have been characterized by several techniques revealing a direct relationship between crystallinity, PEO length and permeability properties. Results at temperatures lower than the Tm of the polyether blocks confirm that lower PEO crystallinity corresponds to higher permeability. At temperatures higher than the Tm of the PEO block, no significant differences were found between the commercial polyamides and the synthesized polyimides. This confirms that the aliphatic phase controls the separation while the hard block provides mechanical strength. Remarkable are the results for the CO2/N2 separation. These new copolyimides are promising materials for post-combustion processes.
Introduction
The rise of carbon dioxide concentration in the atmosphere due to the use of fossil fuels is believed to be a reason for a global climate change which, because of its potentially dangerous effects, requires energetic action. 60% of total CO2 emission is produced by power generation facilities and industrial factories. Of course, an important amount of research should focus on the development of clean energy sources and on more efficient uses of energy. However, it is also necessary to reduce CO2 emission levels from existing industrial sources, so carbon capture and storage (CCS) must be considered as an urgent issue. CO2 can be captured by a variety of methods, which can be classified as post-combustion, pre-combustion and oxy-combustion. Among these methods, post-combustion appears to be one of the most attractive and ready for use alternatives. This process can be applied to different sources: power, steel, cement or petrochemical plants. In this process, the CO2 of the flue gas must be concentrated, purified and compressed (liquefied) to meet the transport and storage specifications. The amount of CO2 in the flue gas ranges from low (4%) to high (30%) concentrations and therefore the technology under development should consider these differences. Other gaseous compounds in the flue gas are H2O, O2, H2, CO, NOx or SOx, although the most frequent is N2. One of the most interesting options for post-combustion CCS are widespread coal fired power plants, where CO2 concentrations are typically around 15 vol%.Nowadays, the role of polymeric membranes applied to gas separation is more and more important. Although a number of them have already an application in industrial separations,1 research is still necessary to discover new materials and/or to improve the properties of existing polymers to ensure actual applicability at an industrial level. In order to guarantee a real application of new polymeric materials in gas separation, an adequate balance of high permeability and good selectivity must be achieved.2,3 Moreover, in addition to the criteria of permeability and selectivity, polymers to be used in new gas separation membranes must have good mechanical properties and thermal resistance to ensure the stability of approximately 100 nm thick defect free selective layers at the required operating conditions.
In order to be useful in such gas separation applications, a polymer film should show a preferential affinity for condensable gases such as CO2 or CH4 as compared with a mostly inert gas such as N2. The polar ether oxygen of poly(ethylene oxide) PEO presents this kind of preferential interaction with carbon dioxide while the interaction with nitrogen molecules is weaker. It was determined that a greater amount of polar ether oxygen groups increases the performance of the polymers in membranes for post-combustion separation processes.4 In this sense, (PEO) compounds give excellent results for the CO2 separation from other light gases due to the existence of this favorable electronic interaction with the ether moiety.5–7
Commercially available multiblock copolymers like PEBAX®, consisting of an alternating sequence of flexible polyether and rigid polyamide blocks, are one of the most popular strategies for CO2/N2 separation. The commercial polymers of the PEBAX® family showed the most promising results for this application. PEBAX® properties can be tailored by varying the chemical structures and the contents of polyamide (PA) and polyether.8 The properties could be improved by the incorporation of different fillers or blending with another oligomer or polymer.9–11 As a function of the composition, there are several kinds of commercially available PEBAX®, but from the literature the best results for the separation of carbon dioxide and nitrogen were found for PEBAX® MH 1657.12–14 The composition of this polymer is 40% semicrystalline amide domains and 60% soft amorphous segments.
On the other hand, copoly(ether–imide)s, containing microphase-separated PEO and imide domains, are of the most interest as membrane selective layer compounds for post-combustion applications. Polyimides are well known for their excellent thermal and oxidative stability, good organic solvent resistance and exceptional mechanical properties, along with an extraordinary ability to separate complex mixtures of gases in diverse applications.2 Typically these materials present a high selectivity but seldom with sufficiently high permeability.15,16 Therefore, it would be convenient to increase permeability without losing selectivity utilizing the ability of copoly(ether–imides) to transport gas molecules through both amorphous domains in contrast to copoly(ether–amide)s where one domain is often a barrier for gas transport due to its semicrystallinity. One of the most common approaches to meet these requirements is the use of aromatic–aliphatic block-copolymers including aliphatic chains showing a certain affinity for condensable gases such as CO2 or CH4. In consequence, block-copolyimides combining aromatic blocks and PEO appear to be a successful route.17,18
In copoly(ether–amide)s and copoly(ether–imide)s, the hard block can be formed by a polymer with well-packed and more or less rigid structures; the soft block, which consists of a polymer with more flexible chains, can form rubbery segments within the polymer chain normally with high free volume and mobility. When aromatic–aliphatic block-copolymers are phase-separated, the glassy segments in the polymer would usually provide mechanical support. The rubbery segments, due to the nature of the flexible chain structure, allow an efficient transport of gas, giving a good permselectivity to the copolymer for the CO2/N2 gas pair19–21 and the amount of these soft segments in the copolymers has a strong influence on the mechanical and gas transport properties.22 This is attributed mainly to the high solubility–selectivity,23 which is due to the strong interactions of CO2 with the oxyethylene group in PEO. The interaction between CO2 and PEO has been discussed and used previously for the development of CO2 selective membranes.24–26
For these reasons it is necessary to carry on studies for a better understanding of this type of PEO copolymers, in order to reach optimal structures for specific applications. In particular, here we have focused on the influence of the kind of copolymer, length of the polyether and effect of the crystallinity on the properties for different copolymers with the same amount of poly(ethylene oxide).
Experimental
Chemicals
Dianhydride 3,3′,4,4′-biphenyl tetracarboxylic dianhydride (BPDA) and 3,3′,4,4′- and the diamines 4,4′-oxydianiline (ODA), poly(ethylene glycol) 6000 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 were purchased from Aldrich. BPDA and ODA were purified by sublimation at high vacuum just before use. Poly(oxyethylene) bis(amine) (Jeffamine ED-2003, n = 41), with nominal molecular weight of 2000 g mol−1, was kindly donated by Huntsman® (Holland). Polyethers were dried at 70 °C in vacuum for 5 h and stored in a desiccator under vacuum until use. PEBAX® MH 1657 was purchased from ARKEMA. Anhydrous N-methyl pyrrolidinone (NMP) and methanol were purchased from Sigma–Aldrich Co. All gases used for gas transport properties tests were purchased from Linde AG and Air Liquide and had a purity of at least 4.5.
000 g mol−1 were purchased from Aldrich. BPDA and ODA were purified by sublimation at high vacuum just before use. Poly(oxyethylene) bis(amine) (Jeffamine ED-2003, n = 41), with nominal molecular weight of 2000 g mol−1, was kindly donated by Huntsman® (Holland). Polyethers were dried at 70 °C in vacuum for 5 h and stored in a desiccator under vacuum until use. PEBAX® MH 1657 was purchased from ARKEMA. Anhydrous N-methyl pyrrolidinone (NMP) and methanol were purchased from Sigma–Aldrich Co. All gases used for gas transport properties tests were purchased from Linde AG and Air Liquide and had a purity of at least 4.5.
Synthesis of copoly(ether–imide)s and preparation of the dense films
The samples were synthesized by combination of the aromatic diamine (ODA) with the aromatic dianhydride (BPDA) and polyethers of different length (PEO2000, PEO6000 and PEO10000). The corresponding copoly(ether–imide) was named after the poly ethylene glycol and the length, because the amount of polyether in the copolymer was always the same (57% w/w) with the exception of PEO2000 with around 55% w/w content. Thus, PEO2000 refers to the sample BPDA-PEO2000-ODA with a weight ratio of the aliphatic diamine and the aromatic diamine of 3.5:1 (w/w) into the copolymer. For the commercial polymer PEBAX® MH 1657, the weight content of PEO is 57%,25 and in this way we can compare the synthesized copolymers under the same conditions.
Polyether poly(oxyethylene oxide) (PEOx000) (x mmol), and 4,4′-oxydianiline (ODA) (y mmol) in the corresponding weight ratios (3.5:1) were dissolved in anhydrous NMP (5 mmol (x + y)/10 mL) in a 100 mL three-necked flask purged with argon. Then, the reaction mixture was cooled to 0 °C, and under mechanical stirring a stoichiometric amount of the dianhydride (BPDA) (x + y mmol) was added and the mixture was stirred overnight at room temperature. During this time the dianhydride completely dissolved and the solution reached high viscosity. Fig. 1 shows the chemical structure of the polymers from a general point of view. In the case of the copoly(ether–imide)s, n is 41 (for PEO2000), 134 (for PEO6000), or 225 (for PEO10000). Table 1 records the proportions of PEO and the acronym for each of the polymers synthesized and used in this work.
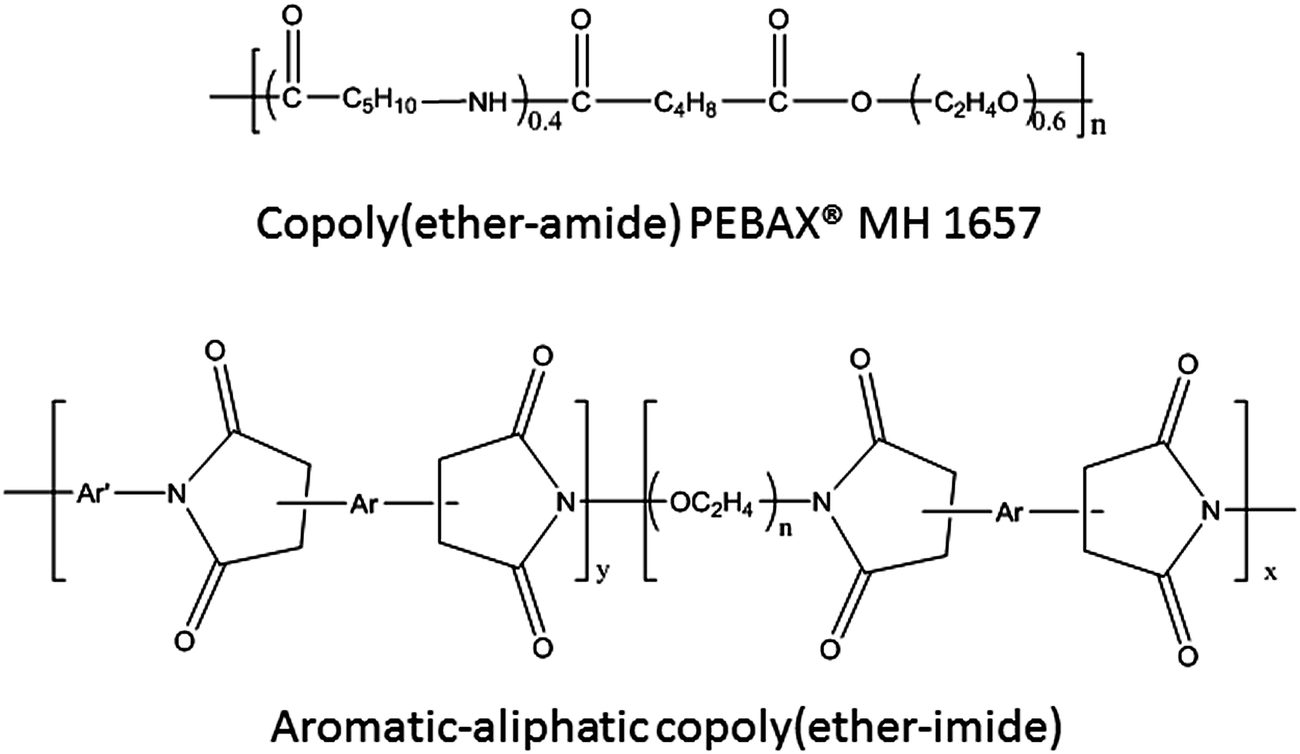 | ||
| Fig. 1 General structures for the copolymers used in this work. | ||
| Copolymer | % PEO theoretical | DSC characterization | ||
|---|---|---|---|---|
| Polyether Tg/°C | Polyether Tm/°C | Crystallinity/% | ||
| PEBAX® ethanol/water | 57 | −50.9 | 15.3 | 19.4 |
| PEBAX® Nmp | 57 | −56.9 | 14.4 | 14.3 |
| PEO2000 | 53.9 | −55.8 | 29.7 | 0.25 |
| PEO6000 | 57.0 | −58.6 | 32.9 | 27.2 |
| PEO10000 | 57.6 | −56.2 | 44.6 | 36.8 |
The resultant viscous polyamic acid solution was diluted with NMP to the appropriate viscosity for casting, filtered through a nominal #1 fritted glass funnel, degassed, and cast onto a leveled glass plate. The resulting film was covered with a conical funnel to avoid fast evaporation of the solvent, dried at 80 °C overnight, and finally treated at 200 °C for 6 hours in a vacuum oven, in order to effect complete solvent evaporation and complete imidization.
For the commercial polymer PEBAX® MH 1657 (from now on, for simplicity, we will refer to it as PEBAX®) the solution was prepared in NMP. After complete dissolution, the same procedure was followed as for other copolymers. After that, thermal treatments under inert atmosphere were carried out at 200 °C. For the preparation of the second PEBAX® membrane the polymer was dissolved at a concentration of 10 wt% in a mixture of ethanol/water (70/30 w/w), with heating at 80–85 °C and magnetic stirring for 24 h. The solution was then filtered and cast at 60 °C for 48 h. Films of the copolymers of 50–70 μm in thickness were obtained. All films showed excellent mechanical properties.
Characterization methods
Attenuated total internal reflectance-Fourier transform infrared analyses (ATR-FTIR) were performed at room temperature using a Bruker ALPHA FT-IR spectrometer in a spectral range of 400–4000 cm−1 with a resolution of 2 cm−1 and an average of 32 scans. The Perkin Elmer Spectrum One infrared spectrometer was equipped with an ATR accessory.A thermal analysis NETZSCH TG209 F1 Iris instrument was used for thermogravimetric study (TGA) of the copolymers and PEBAX®. Disc samples, cut from films, with weights between 5 and 15 mg were tested. This was done with an initial heating rate of 5 K min−1 under a flux of 20 mL min−1 of argon, in a dynamic scan.
Differential scanning calorimetry (DSC) analyses were carried out in a Mettler Toledo (DSC 1 star system) calorimeter equipped with a liquid nitrogen accessory. Disc samples cut from films weighing 5–15 mg were sealed in aluminum pans. Samples were heated at 10 °C min−1 to a target temperature (a “cleaning step” was performed before in order to remove the adsorbed water or solvent traces and the thermal history of the samples).
The permeability, P, was determined by using a permeator with constant volume which uses the time-lag operation method. The measurements were carried out at 700 mbar feed pressure and at thirteen different temperatures from 10 to 70 °C with steps of 5 °C. Although we will focus on CO2 and N2, the permeability was also determined for H2, He, O2 and CH4. A sketch of the device and the analyzed method used has been described in previous studies.27
Results and discussion
Copoly(ether–imide)s imidization
After the films were dried overnight, they were annealed in a vacuum oven at 200 °C for 6 hours to almost complete removal of the solvent. Infrared spectra were recorded to check for the progress of imidization of the copolymers and to compare with the spectra for the PEBAX® samples (Fig. 2).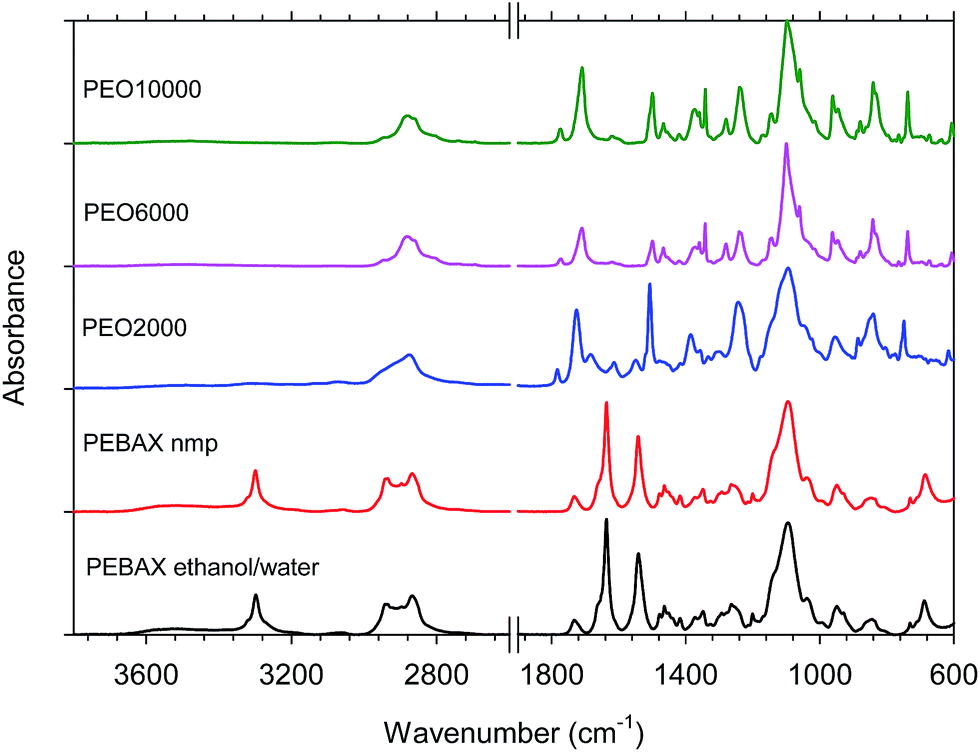 | ||
| Fig. 2 FTIR spectra for the synthetized copolymers and PEBAX® treated at 200 °C. | ||
According to the protocol discussed above, the copolymer films were found to be completely imidized from their FTIR spectra at 200 °C. For the synthesized copolymers, common bands appeared in the spectra: 2895 cm−1 (aliphatic C–H stretching), 1790 cm−1 (symmetric stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O imide groups), 1730 cm−1 (asymmetric stretching of CO imide groups), 1370 cm−1 (C–N stretching of imide groups), 1244 cm−1 (twisting CH2), 1105 cm−1 (C–O stretching), 845 cm−1 (rocking CH2) and 740 cm−1 (imide ring deformation).
O imide groups), 1730 cm−1 (asymmetric stretching of CO imide groups), 1370 cm−1 (C–N stretching of imide groups), 1244 cm−1 (twisting CH2), 1105 cm−1 (C–O stretching), 845 cm−1 (rocking CH2) and 740 cm−1 (imide ring deformation).
In the case of PEBAX®, the characteristic peaks at 1638 cm−1 and 1742 cm−1 represent the presence of O–CO and N–CO bonds. For this commercial copolyamide, common bands appeared in the spectra: 1105 cm−1 (stretching vibrations C–O), 1641 and 1657 cm−1 (stretching of CO imide groups), 3300 cm−1 (vibrations N–H), 2880 cm−1 and 2948 cm−1 (aliphatic C–H stretching). No differences in the spectra were observed between the poly(ether–amide)s dissolved in different solvents.
Thermal stability
Thermogravimetric analysis was performed to evaluate the thermal stability of the polymers. In the case of the copolyimides (annealed at 200 °C for 6 hours), the TGA showed a weight loss pattern consisting of three consecutive steps (see Fig. 3): an initial loss from ambient temperature to 300 °C; a second loss from 300 °C to 460–480 °C; and a third loss from 460–480 °C to 800 °C.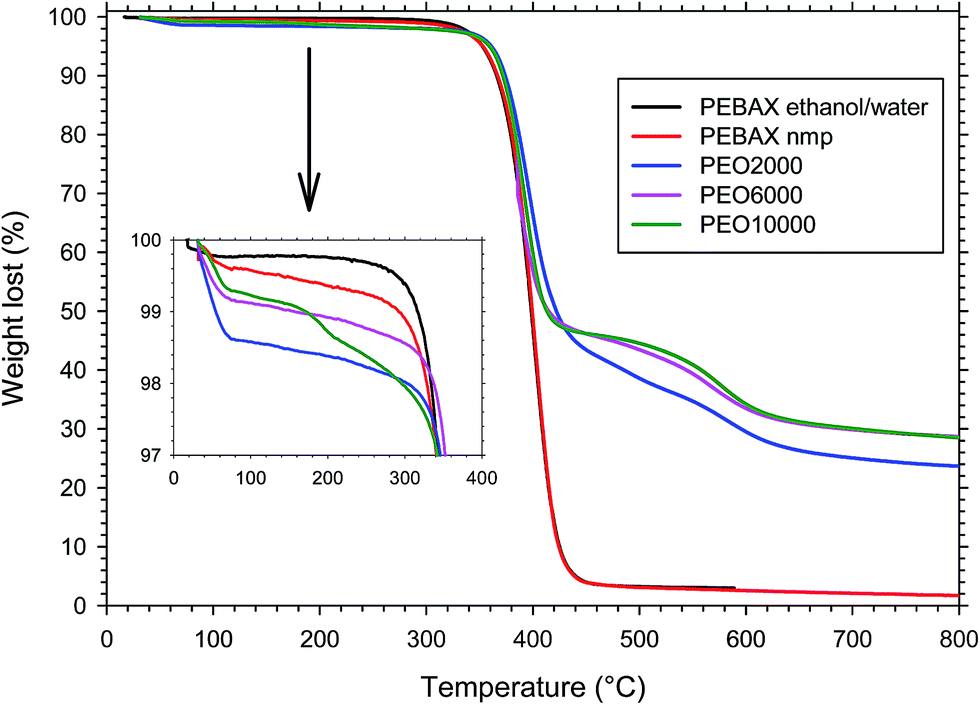 | ||
| Fig. 3 TGA curves in dynamic conditions of copolymers having different poly-ether lengths and PEBAX® annealed at 200 °C for 6 h. | ||
The first loss can be attributed to the absorbed water plus the residual solvent trapped in the film. The weight change for this step was in the order of 2%. The second loss, after correcting for the first one, agreed with the theoretical contribution of PEO entering the copolymer composition,28 and it was therefore assigned to the loss of polyether block sequences. The third and final stage of weight loss was due to the thermal decomposition of the remaining aromatic polyimide segments. TGA analysis confirmed that the polyether chains are much less thermally stable than the aromatic segments, as already found for another copoly(ether–imide) based on PEO23 and therefore a selective degradation of the polyether moiety can be performed in these copolymers. Maximum weight loss rate took place between 360–380 °C for all copolymers.
In the case of the PEBAX® films, although the structure is also made up of hard and soft blocks, like in the synthetized copolymers, the weight loss pattern consisted of two consecutive steps (see Fig. 3): an initial loss from ambient temperature to 100 °C, and a second loss from 300 °C to 460–480 °C. The first loss can be attributed mainly to the absorbed water. The weight change for this step was lower than 1%. The second loss stage was therefore assigned to the thermal decomposition of the polymer. Consequently, for this copoly(ether–amide) the degradation is in one step, showing the same stability for the hard and soft blocks. No differences were observed between the polymers dissolved in ethanol/water or N-methyl pyrrolidinone.
Calorimetric studies
The synthetized copoly(ether–imide)s showed only the Tg, and in some cases the melting temperature, Tm, for the poly(ethylene oxide) segments, and no transition for the aromatic polyimide segments could be detected, but for similar polymers the aromatic Tg was determined at temperatures around 200 °C.22Small changes in the polyether Tg were observed as a function of the PEO length. The melting enthalpy of PEO was taken from the literature as 8.67 kJ mol−1,23 and in this way we could calculate the crystallinity of the aliphatic polyether. A clear tendency was observed in the case of the aliphatic Tm and crystallinity. In the case of the polyimide PEO2000 the crystallinity was almost negligible (0.25%), whereas for the sample PEO6000 the crystallinity was around 27%, and for the longer polyether, PEO10000, the crystallinity was close to 37% (all the values can be found in Table 1). From these data it is clear that longer PEO chains develop higher crystallinities. In the same way, the melting temperature for the polyethers was higher for longer chain lengths.
| Tm PEO10000 (44.6) > Tm PEO6000 (32.9) > Tm PEO2000 (29.7) > PEBAX® (15.3/14.4) |
For PEBAX® samples, the Tm was comparable (15.3 and 14.4 °C) with independence of the solvent. The crystallinity showed some differences between the samples prepared from of ethanol/water or N-methyl pyrrolidinone as a solvent. For ethanol/water the crystallinity was 19.4%, whereas for the samples dissolved in N-methyl pyrrolidinone the crystallinity was 14.3%. This could be related to a better segregation in the samples dissolved in the ethanol/water mixture.
It is well known that gases do not easily pass through crystal structures,29 and for this reason it is important to know the actual crystallinity of our samples as a function of the temperature of permeation measurement. It is necessary to think carefully about the process. In this case, the post-combustion membrane separation process is carried out at pressures close to one bar, and the temperatures generally are between 50–60 °C. Thus, from these results, it can be affirmed that for all the polymers studied in this work, at the operational temperature, the polyether segments will be totally amorphous and above their corresponding Tg.
Gas transport properties
The gas transport properties of the polymers are strongly influenced by the quantity of amorphous or crystalline PEO in the sample. Therefore it is very important to check the permeation performance as a function of the measurement temperature because high temperatures would lower the presence of crystalline phase in the segregated domains that could act as barriers hampering the passage of gases through the film. For this reason, gas transport processes were determined by a two stage procedure used for all the samples under study. The first one is the heating stage, when the separation properties were determined from a low (10 °C) to a high enough temperature for polyether switch from crystalline to an amorphous state (70 °C) with steps of 5 °C between each permeability coefficient measurement temperature, in order to obtain really precise results of the evolution of the samples during the melting process. The second stage is the stepwise cooling with steps of 5 °C, from 70 °C to 10 °C in order to study the crystallization process.From the results of the DSC experiments it was determined that the sample with the highest percentage of crystalline PEO was the sample with the longest one, PEO10000 in this work. The evolution of the CO2 permeability and CO2/N2 selectivity as a function of the measurement temperature for the sample PEO10000 is represented in Fig. 4.
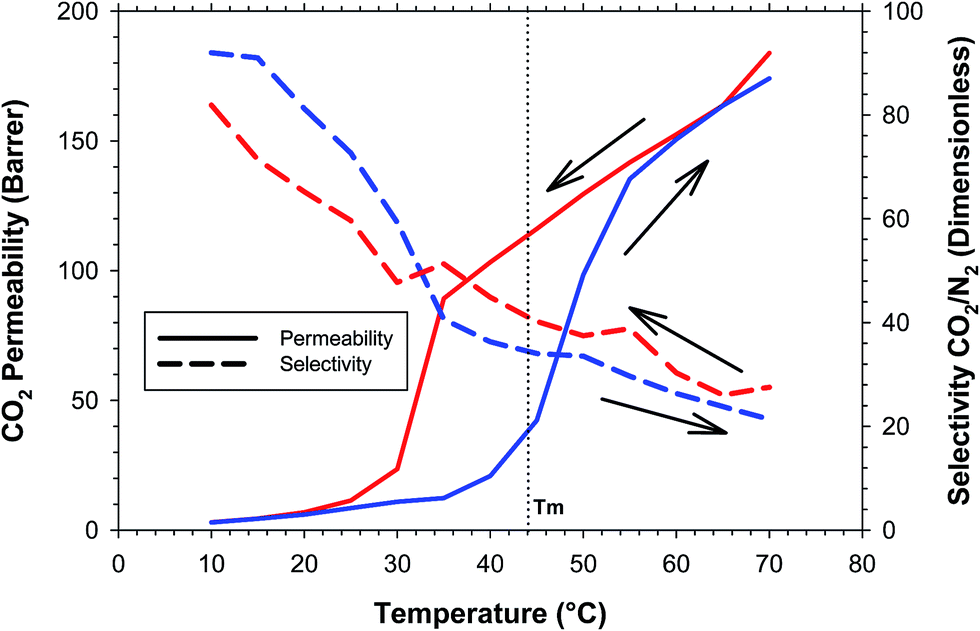 | ||
| Fig. 4 CO2 permeability and CO2/N2 selectivity for the sample PEO10000 as a function of the measurement temperature. | ||
Due the high percentage of crystallinity in the sample (44.6%), one can observe a strong effect of the crystalline phase melting when the sample is heated above the Tm. It is interesting to observe that the rise in permeability starts at temperatures lower than Tm indicating that at temperatures close to Tm an amorphous phase of the PEO starts to act as a plasticizer for macromolecular chains taking part in the crystallite formation. It should be mentioned that the process of the temperature change of the experimental setup is a slow process, the stabilization of the temperature when the tempered part of the setup is heated up or cooled down takes from 1 to 2 h and the results of the permeability measurement at each temperature can be considered as static from the point of view of the temperature influence on the sample in contrast to e.g. DSC or DSC measurements where the sample response to the temperature change is a function of the speed of heat transfer to the sample. This means that at a given temperature of permeability measurement the sample can be considered in an equilibrium state where all parts of the crystalline phase that are able to melt at this temperature are melted. In this sense, during the heating process, it was possible to observe a sharp increase in the permeability at temperatures close to the melting point. Before 35 °C and after 55 °C, the evolution of the permeability coefficient was gradual, as should be expected for the parameters of the thermally activated process. In the range of temperatures 35–55 °C the melting process occurred. During the cooling process, a similar behavior was observed but in this case the range for the crystallization process was between 35 °C to 25 °C. This is a sign that, after membrane “conditioning” by crystallites melting, the behavior of the polymer could be different. With respect to the selectivity, it was not affected significantly by the temperature. Small deviations were observed but the behavior followed the expected path. This observation leads us to the conclusion that the PEO crystalline phase is not taking part in the gas molecule transport through the bulk of a PEO containing polymer and permeability coefficients are increasing and decreasing in accordance with the content of the amorphous phase available for the gas transport.
Similar behavior was observed for the sample PEO6000 but the effect of the crystallinity is lower. During the heating and cooling processes the effect of the melting and crystallization was perceived in the range of 25 to 30 °C. As for the rest of the copolymers, PEO2000 and the PEBAX® membranes (dissolved in methanol/water and dissolved in NMP, respectively), no significant melting and crystallization effects were detected. As in the previous case, no significant changes were observed in the selectivity discarding of course the effect due to the measurement temperature.
Fig. 5 shows the evolution of the CO2 permeability and CO2/N2 selectivity for all the copolymers during the heating process. For lower temperatures the effect of the crystallinity for the PEO6000 and PEO10000 copolymers can be clearly observed. Above the melting temperature the permeability of all the copolymers was comparable. PEO2000 showed a lower permeability than the rest of copolymers, probably due to a slightly lower amount of poly(ethylene glycol) in the copolymer, or because not all the PEO chains, despite being at a similar content, were effective in their interaction with CO2, and this is especially remarkable for PEO with short chain lengths. For the sample of PEBAX® prepared from the solution in NMP the permeability was slightly higher compared to the sample prepared from the ethanol/water solution. The effect could arise from the different solvent quality in respect to PEBAX®, which results in differences of polymer gelation, phase separation etc. during the sample preparation, and differences in the sample microstructure related to the polyamide phase will remain after the thermal treatment procedure used in this work, which does not affect the polyamide phase. The PEO phases in the vicinity of two different hard polyamide phases will behave differently and will result in slight differences e.g. in the CO2 permeability coefficient but not in the selectivity as can be seen from Fig. 5. This effect will be studied in upcoming work. For the rest of the copolymers the results were completely the same.
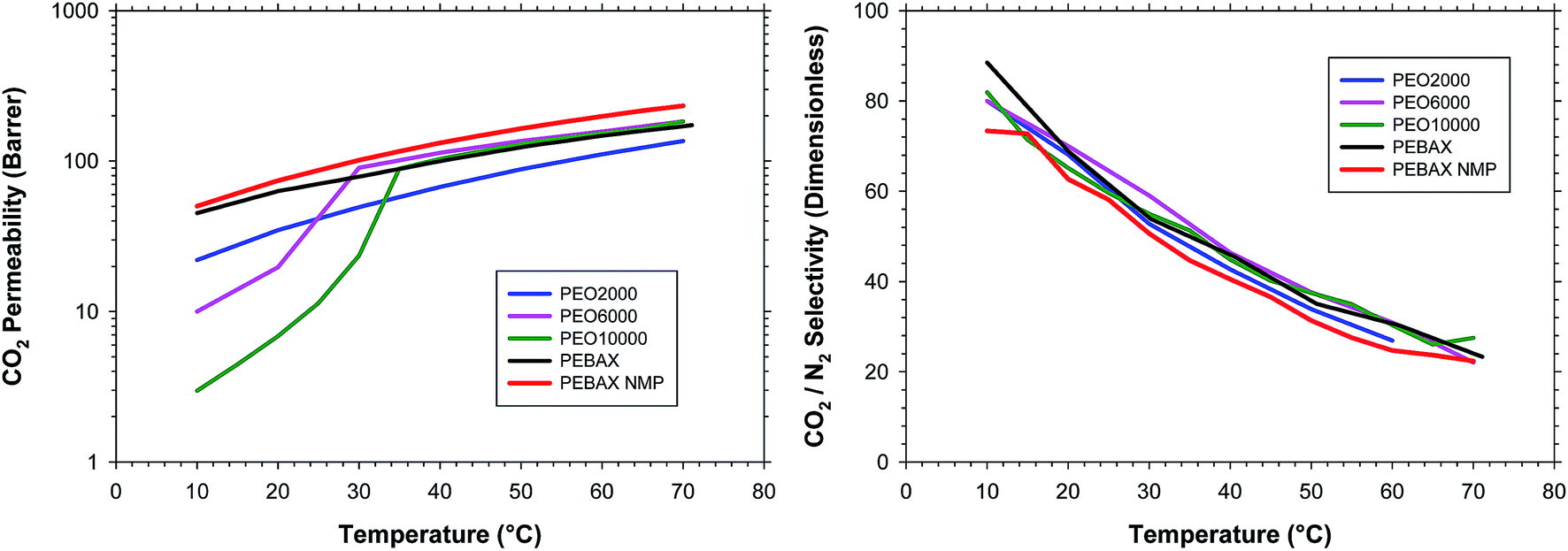 | ||
| Fig. 5 (a) CO2 permeability and (b) CO2/N2 selectivity; as a function of the measurement temperature during the heating process for all the copolymers studied in this work. | ||
Fig. 6 shows a typical Robeson’s plot for the polymers studied. It is possible to observe the evolution of the properties with the measurement temperature, although it should be remembered that the upper boundary was described for an experimental temperature of 35 °C.30 Of course this upper boundary is influenced by the temperature as Rowe et al. confirmed.31 After bypassing the melting point of the polyether, all the samples showed similar behavior. The evolution of the properties with the measurement temperature suggests that the studied materials could bypass Rowe’s upper boundary at higher temperatures.
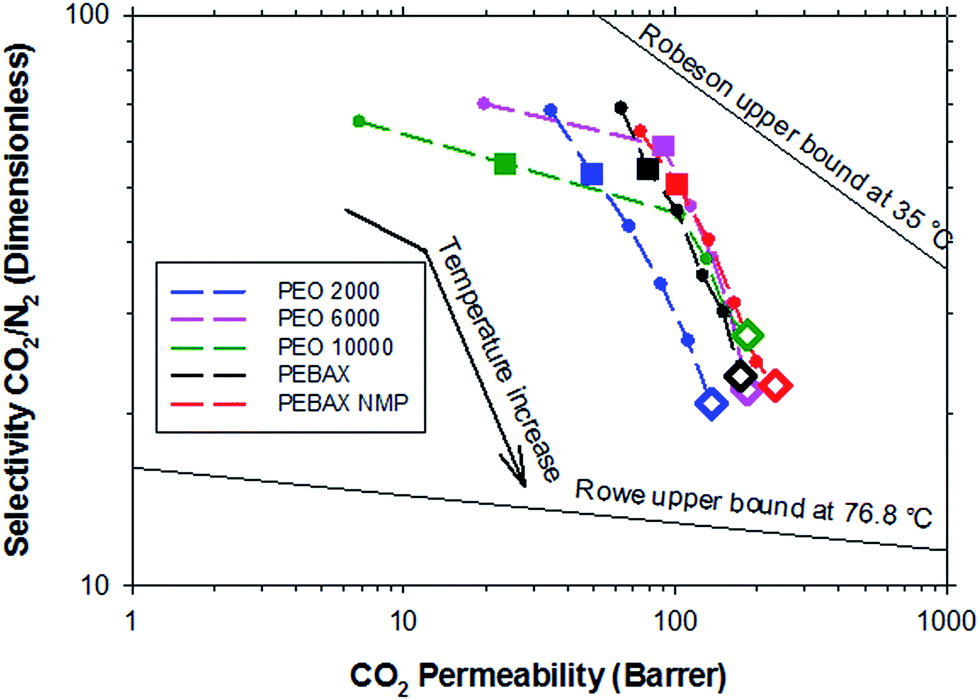 | ||
| Fig. 6 Robeson’s plot for the separation CO2/N2 for the polymers studied at temperatures from 20 to 70 °C (symbols connected with dashed lines). Big filled symbols represent experimental points at 30 °C; big empty symbols at 70 °C. | ||
With regard to the composition of the samples, if we compare poly(ether–amide)s and poly(ether–imide)s the main differences are the amide and imide groups. With respect to the composition of these copolymers, PEBAX® has a linear hard block whereas the copoly(ether–amide)s have a more rigid hard block due to the presence of the crystalline phase. It seems that the effect of the hard block only has a relation to the phase segregation in this kind of copolymers,32 while the separation properties are affected only in the soft segment, poly(ethylene glycol) in both cases. The selectivity is almost the same in all cases, which is clear evidence that the most important aspect is the gas transport through the soft block, while the hard block, being amorphous as in the case of poly(ether–imides), cannot influence significantly the overall gas transport properties due to significantly lower permeability coefficients. After bypassing the melting point, the permeability for all the copolymers became comparable, and again this should be due to the effect of nearly the same content of the soft block in the copolymers under study. This is really important because it is possible to tune materials for post-combustion processes by the simple design of poly(ethylene glycol) copolymers with the most convenient hard block. In this sense, it will be possible to look for an improvement in solvent and thermal resistance and the most adequate solubility of the polymer as long as the copolymers are capable of undergoing phase segregation.
Comparing the permeation properties of the segments22,25,33,34 with the properties of the materials used in this work, it is simple to observe that the main separation abilities are provided by the rubbery segments. Table 2 shows the gas transport parameters for the blocks and for the copolymers. For the copolymer used in this work, two temperatures were described. The first temperature (35 °C) was used to compare with the literature data. The second one (60 °C) was selected for two reasons: it was high enough to have all the poly(ethylene glycol)s in the amorphous state; and it is within the application temperature range of post-combustion processes. Values of permeability coefficient determined at 35 °C are shown for both heating and cooling stages to demonstrate the effect of the PEO recrystallization in copoly(ether–imide)s.
| Polymer | Temperature/°C | Permeability (Barrer) | Selectivity (Dimensionless) | Reference | ||
|---|---|---|---|---|---|---|
| P(H2) | P(CO2) | α CO2/N2 | α CO2/H2 | |||
| a For these samples, at this temperature, there is some degree of crystallinity, for this reason it was recorded the permeability of the heating and cooling cycles. | ||||||
| Nylon 6 | 30 | 0.58 | 0.00049 | 9.6 | 0.00084 | 33 |
| BPDA-ODA | 30 | 3.68 | 0.90 | 38.0 | 0.24 | 22 and 34 |
| PEO semi-crystalline | 35 | 1.8 | 12.0 | 48.0 | 6.7 | 25 |
| PEO amorphous | 35 | 21.0 | 143.0 | 47.7 | 6.8 | 25 |
| PEBAX® 1657 ethanol/water | 35 | 6.67 | 90.1 | 47.3 | 13.5 | This work |
| 60 | 23.6 | 149.7 | 28.6 | 6.3 | ||
| PEBAX® 1657 Nmp | 35 | 13.2 | 116.2 | 44.7 | 8.8 | This work |
| 60 | 32.2 | 198.4 | 24.7 | 6.2 | ||
| PEO2000 | 35 | 7.6 | 54.8 | 47.7 | 7.2 | This work |
| 60 | 19.8 | 111.2 | 26.9 | 5.6 | ||
| PEO6000 | 35a | 10.4/11.3 | 95.8/101.8 | 49.0/51.1 | 9.2/9.0 | This work |
| 60 | 22.9 | 153.0 | 30.4 | 6.7 | ||
| PEO10000 | 35a | 2.7/8.9 | 12.36/89.31 | 40.6/51.3 | 4.6/10.0 | This work |
| 60 | 24.7 | 151.5 | 30.1 | 6.1 | ||
As expected, the permeabilities for the homopolymers or hard blocks (nylon 6 and BPDA-ODA) are low compared to those of PEO, and the selectivities for this glassy polymers were considerably different from each other and from PEO. Considering the crystallites as an impermeable barrier, the amorphous PEO has a permeability one order of magnitude higher than the semicrystalline PEO. Despite this, the selectivity is similar, indicating that the permeation process occurs through the remaining amorphous phase. A more extended study on gas transport mechanisms in the PEBAX® membranes with different polyethylene glycol lengths was carried out by Rahman et al.35 If we analyze the results for the copolymers, we should expect three different possibilities for the evolution of the permeation properties: permeability similar to the hard block, similar to the soft block, or a combination of properties between both segments. The results showed properties closer to the behavior of the soft blocks. This is a clear indication that the gas transport is fully controlled by the properties of the amorphous poly(ethylene glycol) phase. If the process occurs through the PEO, and all the copolymers are present in the same amount, similar properties are expected for all of them. This was found to occur in many but not all the cases and temperatures.
For PEBAX® samples prepared from different solvents significant differences in properties were found. The copolymer dissolved in NMP showed higher permeabilities and lower selectivities than the copolymer dissolved in ethanol/water. Once verified by TGA that no solvent remained in the copolymers, the explanation should be in relation with the interaction between the chains. The elimination of some of the typical hydrogen bonds formed in polyamides could produce an increase in the mobility chains, and then an increase in the permeability most probably with some effect on the selectivity. These effects will be studied in more detail in future work. In the case of the copolyimide, at 35 °C the crystallinity has a really important role. Higher crystallinity, like in the case of PEO10000, produced low permeabilities. Comparing the heating and the cooling cycle for PEO10000, great differences were observed in the selectivities (CO2/N2 and CO2/H2). While the selectivity in the heating cycle was low, during the cooling cycle the selectivity was similar to the other copolyimides, and higher than the selectivity for the copolyamide PEBAX®.
Comparing the synthetized copolymers, the permeability for the sample PEO2000 was lower than for the rest of copolymers. Shorter chains produced lower mobility and then lower permeability. The permeability at 60 °C stood out as interesting. The permeability values were almost the same for the samples PEBAX® (ethanol/water), PEO6000 and PEO10000, and the selectivity was higher for the copoly(ether–imides). Therefore, although it is true that the permeation properties are regulated, especially the permeability, by the soft block, there could be an influence of the hard block on the selectivity factor. The CO2 permeability for the copolymers is not comparable to the permeability of pure amorphous PEO, but of course the content of the copolymers is around 57% while the pure PEO is 100%. The permeability could be increased in an effective way by for example the addition of inorganic nanoparticles.36,37 Nevertheless, as effectively predicted,38 the evolution in the permeability should not result in a really large growth in permeability. On the other hand, a certain level of environmental resistance and mechanical properties is necessary, which is not simple when the PEO content is very high due mainly to the high solubility in solvents like water, and very poor mechanical properties.
The dependence of gas diffusion on temperature can be expressed in terms of an Arrhenius type relationship that considers the movement of the gas molecules through a membrane as a thermally activated process.39 Therefore, by an Arrhenius representation, it was possible to obtain the activation energies for the carbon dioxide and nitrogen permeability (Ep) (displayed in Table 3).
| PEBAX® EtOH/H2O | PEBAX® Nmp | PEO2000 | PEO6000 | PEO10000 | |
|---|---|---|---|---|---|
| Ep/kJ mol−1 | 13.3 | 16.1 | 20.8 | 14.4 | 16.2 |
| EpCO2 − EpN2/kJ mol−1 | −17.1 | −18.2 | −21.2 | −17.8 | −18.3 |
The positive or negative character of the energies has a direct relationship with the evolution of the behavior with the temperature. In this sense, the CO2 permeability will increase with the temperature, and the bigger increase should be observed in the sample PEO2000. In the case of the selectivity, the behavior is opposite as tested previously. With the exception of PEO2000, the rest of the samples showed very close values, so a similar evolution of separation properties of the samples with the temperature can be expected.
Conclusions
A set of new copoly(ether–imide)s were prepared by the reaction between an aromatic dianhydride (BPDA), an aromatic diamine (ODA), and poly(ethylene oxide) diamine (PEO) with different length (2000, 6000 and 10000) and the same content, in order to compare with the commercial copoly(ether–amide) PEBAX® MH 1657 (obtaining one from methanol/water and the other from N-methyl pyrrolidinone). All the copolymers are able to experience phase segregation, where the aromatic phase contributed to the mechanical and thermal stability of the polymer, and the aliphatic phase contributed to the high CO2 permeability.
For copolyimides, the thermal stability in inert atmosphere was above 350 °C, showing two-step decomposition, as the PEO segments are the less thermally stable units. For copolyamides, the thermal analysis showed one-step decomposition at similar temperatures.
By calorimetric techniques the Tg, Tm and crystallinity of the copolymers were determined. PEO Tg values between −50 and −57 °C were found for all the samples. For PEBAX® samples small differences were found for the Tm and crystallinity, showing in both cases higher values than the sample obtained from the mixture methanol/water. Copolyimides showed an evolution with the molecular weight of the PEO. Then at higher PEO lengths higher Tm and higher crystallinity were found.
Different heating and cooling cycles were carried out in order to determine the influence and evolution of the crystallinity with the permeability measurement temperature. No differences in the permeability properties of the samples were observed at temperatures where the amount of crystalline PEO was zero or negligible (>50 °C). With the exception of the shorter polyethylene glycol (2000 g mol−1), the copolyimides showed similar results than the commercial copolyamide PEBAX®. It is therefore possible to tune the structure of the polymers keeping the same properties.
These results show that it is possible to obtain PEO copolymers with controlled structures and really promising results for applications in post-combustion processes. Thus, the properties can be tuned to some extent by simply varying the aromatic composition of the copolymer. Besides, it is possible to control several parameters: cost, processability, solubility and other factors in order to adapt these copolymers for other specific applications and processes.
Acknowledgements
The authors would like to thank Silvio Neuman and Judith Grünauer for technical support. This work was financially supported by the Helmholtz Association of German Research Centers through the Helmholtz Portfolio MEM-BRAIN.Notes and references
- R. W. Baker, Ind. Eng. Chem. Res., 2002, 41, 1393–1411 CrossRef CAS.
- Polyimides: Thermally Stable Polymers, ed. M. T. Bessonov, M. M. Koton, V. V. Kudryavtsev and L. A. Laius, Consultants Bureau, New York, 1987 Search PubMed.
- E. Favre, J. Membr. Sci., 2007, 294, 50–59 CrossRef CAS PubMed.
- A. Tena, A. Marcos-Fernández, L. Palacio, P. Cuadrado, P. Prádanos, J. de Abajo, A. E. Lozano and A. Hernández, Ind. Eng. Chem. Res., 2012, 51, 3766–3775 CrossRef CAS.
- H. Z. Chen, Y. C. Xiao and T.-S. Chung, J. Membr. Sci., 2011, 381, 211–220 CrossRef CAS PubMed.
- Y. Hirayama, Y. Kase, N. Tanihara, Y. Sumiyama, Y. Kusuki and K. Haraya, J. Membr. Sci., 1999, 160, 87–99 CrossRef CAS.
- M. Minelli, M. Giacinti Baschetti, D. T. Hallinan Jr and N. P. Balsara, J. Membr. Sci., 2013, 432, 83–89 CrossRef CAS PubMed.
- S. L. Liu, L. Shao, M. L. Chua, C. H. Lau, H. Wang and S. Quan, Prog. Polym. Sci., 2013, 38, 1089–1120 CrossRef CAS PubMed.
- J. Lillepärg, P. Georgopanos and S. Shishatskiy, J. Membr. Sci., 2014, 467, 269–278 CrossRef PubMed.
- M. M. Rahman, V. Filiz, S. Shishatskiy, C. Abetz, S. Neumann, S. Bolmer, M. M. Khan and V. Abetz, J. Membr. Sci., 2013, 437, 286–297 CrossRef CAS PubMed.
- A. Car, C. Stropnik, W. Yave and K.-V. Peinemann, J. Membr. Sci., 2008, 307, 88–95 CrossRef CAS PubMed.
- V. Bondar, B. Freeman and I. Pinnau, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 2051–2062 CrossRef CAS.
- B. Wilks and M. E. Rezac, J. Appl. Polym. Sci., 2002, 85, 2436–2444 CrossRef CAS.
- J. H. Kim, S. Y. Ha and Y. M. Lee, J. Membr. Sci., 2001, 190, 179–193 CrossRef CAS.
- D. Ayala, A. E. Lozano, J. de Abajo, C. García-Perez, J. G. de la Campa, K. V. Peinemann, B. D. Freeman and R. Prabhakar, J. Membr. Sci., 2003, 215, 61–73 CrossRef CAS.
- K. Tanaka, H. Kita, K. Okamoto, A. Nakamura and Y. Kusuki, J. Membr. Sci., 1989, 47, 203–215 CrossRef CAS.
- K.-i. Okamoto, M. Fujii, S. Okamyo, H. Suzuki, K. Tanaka and H. Kita, Macromolecules, 1995, 28, 6950–6956 CrossRef CAS.
- H. Suzuki, K. Tanaka, H. Kita, K. Okamoto, H. Hoshino, T. Yoshinaga and Y. Kusuki, J. Membr. Sci., 1998, 146, 31–37 CrossRef CAS.
- T. A. Barbari, W. J. Koros and D. R. Paul, J. Membr. Sci., 1989, 42, 69–86 CrossRef CAS.
- A. Tena, A. E. Lozano, L. Palacio, A. Marcos-Fernández, P. Prádanos, J. de Abajo and A. Hernández, Int. J. Greenhouse Gas Control, 2013, 12, 146–154 CrossRef CAS PubMed.
- D. Husken, T. Visser, M. Wessling and R. J. Gaymans, J. Membr. Sci., 2010, 346, 194–201 CrossRef CAS PubMed.
- A. Tena, A. Marcos-Fernández, L. Palacio, P. Prádanos, A. E. Lozano, J. de Abajo and A. Hernández, J. Membr. Sci., 2013, 434, 26–34 CrossRef CAS PubMed.
- A. Tena, A. Marcos-Fernández, A. E. Lozano, J. G. de la Campa, J. de Abajo, L. Palacio, P. Prádanos and A. Hernández, J. Membr. Sci., 2012, 387–388, 54–65 CrossRef CAS PubMed.
- A. Car, C. Stropnik, W. Yave and K. V. Peinemann, Sep. Purif. Technol., 2008, 62, 110–117 CrossRef CAS PubMed.
- H. Lin and B. D. Freeman, J. Membr. Sci., 2004, 239, 105–117 CrossRef CAS PubMed.
- S. R. Reijerkerk, M. H. Knoef, K. Nijmeijer and M. Wessling, J. Membr. Sci., 2010, 352, 126–135 CrossRef CAS PubMed.
- A. M. Shishatskii, Y. P. Yampol’skii and K. V. Peinemann, J. Membr. Sci., 1996, 112, 275–285 CrossRef CAS.
- A. Marcos-Fernández, A. Tena, A. E. Lozano, J. G. De La Campa, J. De Abajo, L. Palacio, P. Prádanos and A. Hernández, Eur. Polym. J., 2010, 46, 2352–2364 CrossRef PubMed.
- C. Casado-Coterillo, J. Soto, M. T. Jimaré, S. Valencia, A. Corma, C. Téllez and J. Coronas, Chem. Eng. Sci., 2012, 73, 116–122 CrossRef CAS PubMed.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS PubMed.
- B. W. Rowe, L. M. Robeson, B. D. Freeman and D. R. Paul, J. Membr. Sci., 2010, 360, 58–69 CrossRef CAS PubMed.
- A. Tena, A. Marcos-Fernández, A. E. Lozano, J. G. de la Campa, J. de Abajo, L. Palacio, P. Prádanos and A. Hernández, J. Membr. Sci., 2012, 387–388, 54–65 CrossRef CAS PubMed.
- E. Picard, A. Vermogen, J. F. Gérard and E. Espuche, J. Membr. Sci., 2007, 292, 133–144 CrossRef CAS PubMed.
- J. Y. Park and D. R. Paul, J. Membr. Sci., 1997, 125, 23–39 CrossRef CAS.
- M. M. Rahman, V. Filiz, S. Shishatskiy, C. Abetz, P. Georgopanos, M. M. Khan, S. Neumann and V. Abetz, ACS Appl. Mater. Interfaces, 2014 DOI:10.1021/am504223f.
- V. Nafisi and M.-B. Hägg, J. Membr. Sci., 2014, 459, 244–255 CrossRef CAS PubMed.
- V. Nafisi and M.-B. Hägg, ACS Appl. Mater. Interfaces, 2014, 6, 15643–15652 CAS.
- A. Tena, M. de la Viuda, L. Palacio, P. Prádanos, Á. Marcos-Fernández, Á. E. Lozano and A. Hernández, J. Membr. Sci., 2014, 453, 27–35 CrossRef CAS PubMed.
- R. E. F. Kesting, Polymeric gas separation membranes, John Wiley & Sons, New York, 1993 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |