
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzymatic multi-functionalization of microparticles under aqueous neutral conditions†
G. Faccio*,
S. Senkalla,
L. Thöny-Meyer and
M. Richter
Empa, Swiss Federal Laboratories for Materials Science and Technology – Laboratory for Biointerfaces, Lerchenfeldstrasse 5, 9014 St. Gallen, Switzerland. E-mail: greta.faccio@empa.ch
First published on 16th February 2015
Abstract
The immobilization of proteins and enzymes on microparticles enables innovative biofunctionalization and facilitates the recycling of, for example, catalysts in a biocatalytic process. When enzymes are used for the immobilization of proteins to material surfaces, site-specificity can be achieved, and the immobilization reaction can proceed under mild reaction conditions in an aqueous environment. By selectively oxidizing the exposed tyrosyl side chains of protein to reactive o-quinones using molecular oxygen as electron acceptor, tyrosinase promotes the formation of new covalent bonds via 1,4-addition of nucleophilic moieties to the o-quinones. The introduction of tyrosinase-susceptible tyrosine residues (Y-tag) to a protein by genetic engineering can thus enable site-specific crosslinking and site-specific protein immobilization. In this study, several variants of the fluorescent protein GFPuv were produced in order to investigate the tyrosinase-mediated crosslinking and immobilization reaction. The Y-tag was a target for rapid protein–protein crosslinking by tyrosinase catalysis. Moreover, low concentrations of tyrosinase were sufficient to obtain detectable fluorescent microparticles using the Y-tagged GFPuv variants, e.g. 0.4 μM (50 μg ml−1) protein concentrations. Eventually, we showed that this enzyme-based technology allows for the multiple functionalization of microparticles using different fluorescent proteins.
Introduction
Biohybrid devices combining inorganic substrates and biomolecules are increasingly finding applications in diverse fields spanning from medical biosensing, biocatalysis, point-of-care monitoring, to solar energy conversion. Especially when multi-enzyme cascade reactions are necessary, the optimal immobilization of all catalysts is crucial. Immobilization of desired biomolecules is often achieved either by simple adsorption (non-covalent), by entrapment during fabrication, or by wet chemical modification using chemical crosslinkers (covalent), and it often proceeds through a trial and error approach.1In a biomimetic approach, crosslinking enzymes are attracting attention as biocatalysts for protein immobilization as they can provide high selectivity for site-specific modifications. Additionally, as catalysts they are not consumed during the crosslinking reaction and thus reusable, and they can be produced in a sustainable biotechnological manner. Crosslinking enzymes such as the copper-dependent tyrosinase (EC 1.14.18.1) promote the formation of new covalent bonds between proteins carrying the target residue. By selectively and consecutively hydroxylating and oxidizing tyrosyl groups to the corresponding o-quinone, tyrosinase can introduce novel Tyr–Tyr, Tyr–Cys, Tyr–Lys, and Tyr–His bonds.2 Tyrosinase from mushroom is commercially available and safely used as additive in food applications.3 In green chemistry, it was recently used directly from the crude mushroom extract for the synthesis of annulated benzofuranes in a domino reaction with laccase.4 The crosslinking activity of tyrosinase has been used to develop in situ forming hydrogels,5 to modify the structure of complex protein-based networks,6 and to stabilize emulsions.7,8
For most crosslinking enzymes, however, the accessibility of the target residues on a substrate protein on the substrate molecule is the limiting factor for their action. In the case of tyrosinase, the hydrophobicity of tyrosine residues often confines these residues to the interior of protein structures making them inaccessible to the enzymes.9 Thus, crosslinking of well-folded proteins by these enzymes is not always possible. This apparent limitation of substrate accessibility for tyrosinase might however, be overcome when combined with protein engineering strategies. For example, the addition of genetically encoded additional tyrosines to external flexible regions of a substrate protein might provide a novel recognition site for tyrosinase. Since a high number of proteins carries both N- and C-termini exposed on the surface10 these represent optimal sites for protein engineering in this respect. Few studies have investigated the use of tyrosine-containing tags for protein crosslinking.11,12 We recently reported the site-specific immobilization of phycocyanin with tyrosinase to a solid surface through a 21-amino acid long N-terminal tag containing two tyrosine residues.13 Microparticles have found application in various fields such as medicine, biochemistry, and colloid chemistry, especially after being appropriately functionalized.14 Various synthetic approaches have been developed for protein immobilization15 in both covalent and non-covalent ways16 to produce fluorescent particles. Here we report the production of fluorescent microparticles under aqueous neutral conditions using minimal amounts of catalyst, e.g. the enzyme tyrosinase, that is not incorporated into the final product and thus reusable. Specifically, we used engineered forms of GFPuv17 and C-phycocyanin to investigate crosslinking with tyrosinase, to establish the conditions enabling the optimal immobilization of proteins, and to produce multi-functionalized microparticles carrying two different proteins.
Experimental
Materials, proteins, and software
Tyrosinase from mushroom (T3824, ≥1000 unit per mg), and L-tyrosine were purchased from Sigma Aldrich (Buchs, Switzerland). The tyrosinase preparation used in this study had an activity and specific activity on L-tyrosine under the tested conditions of ∼25 nkatal ml−1 and 12 nkatal mg−1, respectively. Restriction enzymes were from New England BioLabs (UK). The α-subunit of C-phycocyanin (HisCPC) was recombinantly produced in E. coli as reported in ref. 13. Chromatographic media and columns were from GE Healthcare (Switzerland). Amino-modified polystyrene microparticles were purchased from Polysciences Inc., (Warrington, USA). Ultrapure water was utilized in the preparation of the solutions used in this study. The software ImageJ18 was used for the densitometric analysis of images of SDS polyacrylamide gels, e.g. band intensity and molecular weight calculation. The software Pymol (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC) was used for analysis of the three dimensional structure of GFPuv and tyrosinase that were retrieved from the Protein Data Bank (PDB; http://www.rcsb.org/pdb/).Genetic engineering of GFPuv
Plasmid pPP1 harbouring the genes coding for the C-terminally His-tagged version of GFPuv and conferring ampicillin resistance was used as template for the construction of the GFPuv variants.19 Oligonucleotides were designed for cloning by isothermal assembly.20 In order to produce C-terminally Tyr-tagged GFPuv, codons for the one- and five-tyrosine tags were inserted after the His-tag coding region into the plasmid by PCR using Phusion® High-Fidelity DNA Polymerase (New England Biolabs), the plasmid pPP1 and oligonucleotides as in ESI S1.† After incubation for 15 min at 50 °C, 1 μl of reaction was used to transform chemically competent E. coli DH5α cells. Constructs were verified by sequencing and used to transform the production strain E. coli BL21(DE3). Mutagenesis of GFPuv and Y1-GFPuv was performed with the Quikchange Lightning Multi-Site Directed mutagenesis kit (Agilent Technologies) and the mutagenic oligonucleotides as in Table S1.† As before, E. coli strain DH5α was used for DNA manipulation steps, and plasmids confirmed by sequencing were transformed into the production strain E. coli BL21(DE3).Expression of GFPuv variants in E. coli
Few microliters of the glycerol stock of E. coli BL21(DE3) strains previously transformed with the appropriate plasmid were used to inoculate a pre-culture of 100 ml LB medium supplemented with ampicillin (0.1 mg ml−1). After overnight incubation at 37 °C, 160–180 rpm, this was used to inoculate 2 l cultures containing 500 ml of selective LB medium with an initial OD600 of 0.04. At an OD600 of ∼0.5, IPTG was added to a final concentration of 1 mM to induce expression, and cultivation was proceeded at 30 °C at 180 rpm for approximately 20 h. Cells were harvested by centrifugation and stored at −20 °C.Purification of GFPuv variants by affinity chromatography
Purification of the GFPuv variants started by incubating the biomass in the presence of lysozyme (0.1 mg ml−1) in 100 mM K-phosphate pH 6.8 for 30 min on ice (5 ml per g cells). After a freeze–thaw step at −80 °C, Benzonase® solutions (250 U per g cells) was added following incubation at 37 °C shaking for 1 h. Sonication for 12 cycles at 80% power with 10 s intervals on ice and centrifugation at 4 °C at 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 808 g (SS34 rotor, Sorvall centrifuge) allowed the removal of the cell debris and the recovery of the cell-free extract. All GFPuv variants were purified by immobilized metal ion affinity chromatography (IMAC) on Histrap columns using 100 mM K-phosphate pH 7 buffer and by removing non-specifically bound E. coli proteins in a step-wise manner at 50 and 100 mM imidazole concentration, respectively. GFPuv variants eluted at an imidazole concentration of 300 mM and, after the greenish fractions were pooled, the imidazole was removed using a Vivaspin ultrafiltration device with a molecular weight cut-off of 10000 (Sartorius, Switzerland). Protein concentration was determined either by the Bradford method using bovine serum albumin as standard, or using the extinction coefficient 27000 M−1 cm−1 and a calculated molecular mass of 27862 g mol−1.
808 g (SS34 rotor, Sorvall centrifuge) allowed the removal of the cell debris and the recovery of the cell-free extract. All GFPuv variants were purified by immobilized metal ion affinity chromatography (IMAC) on Histrap columns using 100 mM K-phosphate pH 7 buffer and by removing non-specifically bound E. coli proteins in a step-wise manner at 50 and 100 mM imidazole concentration, respectively. GFPuv variants eluted at an imidazole concentration of 300 mM and, after the greenish fractions were pooled, the imidazole was removed using a Vivaspin ultrafiltration device with a molecular weight cut-off of 10000 (Sartorius, Switzerland). Protein concentration was determined either by the Bradford method using bovine serum albumin as standard, or using the extinction coefficient 27000 M−1 cm−1 and a calculated molecular mass of 27862 g mol−1.
Enzymatic activity measurements
A stock solution of tyrosinase was dissolved at a 1–2 mg ml−1 concentration in 100 mM K-phosphate pH 6.8. Tyrosinase activity measurements were performed by adding 10 μl of tyrosinase solution to 190 μl of 2 mM L-tyrosine prepared in 100 mM K-phosphate pH 6.8 in a 96-well plate and monitoring the variation of absorbance at 475 nm at 22 °C. Activity values were calculated by using a 3600 M−1 cm−1 extinction coefficient for dopachrome at 475 nm, and a 0.56 constant value to correct for the path length. Tyrosinase activity on GFPuv variants was measured by adding 10 μl of tyrosinase solution to 190 μl of diluted GFPuv variant at 22 °C in the same concentrations used for protein immobilization and monitoring absorbance at 290 nm and 475 nm over time. Possibly contaminating laccase activity in the tyrosinase solution was measured under identical conditions using 100 mM K-phosphate pH 6.8 and 5 mM ABTS by monitoring changes in absorbance at 420 nm overtime. All reactions were performed in triplicate. Activity measurements were performed using a Cary Varian Bio spectrophotometer and 96-well UV-transparent plates.Crosslinking of GFPuv variants in solution using tyrosinase
Protein crosslinking was carried out by incubating the substrate protein, e.g. GFPuv variants, and a variable amount of tyrosinase in 100 mM K-phosphate at pH 6.8 and at 22 °C for a certain time. Crosslinking reactions were analysed by SDS PAGE with in-house made 16% acrylamide gels, after incubation for 5 min at 95 °C, and stained with Coomassie (GelCode Blue Stain Reagent, Pierce). All reactions were performed in triplicate. The molecular weight of the reaction products of the relevant protein bands was calculated using a calibration curve obtained by plotting the migration distance in denaturing SDS polyacrylamide gels and the MW of the MW standards used for each gel (Fig. S2†). When samples contained HisCPC, the gel was also stained for bilin-containing proteins using 100 mM zinc acetate for 5 min at room temperature and imaged under UV light with a 15 s exposure time.Functionalization of microparticles using tyrosinase
Immobilization of GFPuv variants and HisCPC to microparticles was performed by first transferring the microparticles contained in 10 μl of the stock solutions (1.68 × 109 particles per ml, Polybead® Amino Microspheres, diameter 3 μm, Polysciences, Inc., Eppelheim, Germany) to the appropriate buffer, i.e. 100 mM K-phosphate pH 6.8, by repeated centrifugation and resuspension in buffer. Equilibrated microparticles were incubated with the substrate protein, e.g. a selected GFPuv variant or HisCPC, and tyrosinase in a 50 μl final volume in 100 mM K-phosphate pH 6.8. The effect of the tyrosinase concentration was tested by incubating 10 μl of pre-equilibrated microparticles with tyrosinase in a 0–4 mg ml−1 range and 0.5 mg ml−1 GFPuv variant for 3 h at 22 °C. The influence of the GFPuv concentration was tested by incubating 10 μl of pre-equilibrated microparticles with 0.05 mg ml−1 tyrosinase and each GFPuv variant at a concentration of 0–5.5 mg ml−1 for 3 h at 22 °C. Co-immobilization of Y1-GFP and HisCPC was similarly performed using each a 0.5 mg ml−1 HisCPC and Y1-GFP concentration, respectively, plus adding 0.05 mg ml−1 tyrosinase prior to incubation for 3 h at 22 °C. After harvesting by centrifugation and removal of the unbound proteins in the supernatant, the microparticles were rinsed three times by resuspending them in 100 μl of buffer. In all experiments, microparticles incubated with the GFPuv variant or HisCPC in the absence of tyrosinase was used as a reference to establish the extent of adsorption to the surface. All reactions were performed in triplicate and carried out in BRAND® 96-well PCR plate (Sigma Aldrich, Switzerland) in order to facilitate handling.Analysis of the functionalized microparticles by flow cytometry
For flow cytometry analysis, 3–10 μl of microparticles solution was added to 1 ml of ultrapure water, quickly vortexed, and measured with a CyFlow flow cytometer equipped with a blue laser diode (10 mW) at 473 nm (Partec, Germany). Samples were analysed setting the forward scattering (FSC) signal as trigger with an associated gain of 250, and a lower threshold (L-L) of 0. For the green fluorescence signal (FL-1), a gain of 600 and a L-L of 10 were set. The flow rate was 6 μl s−1. The fluorescence of the microparticle population was indicated by GMn-x value (geometric mean of FL-1 fluorescence intensity) calculated by the software provided with the instrument (Partec, Germany).Analysis of the functionalized microparticles by fluorescence microscopy
Microparticles were also visually analysed with an inverted microscope Leica DM6000 fitted with a digital camera (Leica DM6000, Wetzlar, Germany) and equipped with a GFP (λex = 450–490 nm, λem = 525–550 nm) and N2.1 filter (λex = 515–560 nm, λem = >590 nm). For the acquisition of fluorescence images, exposure, gain, and intensity parameters were kept constant within each experiment in order to allow comparison of the samples. Confocal laser scanning microscopy (CLSM) of microparticles was carried out using a LSM 780 from Carl Zeiss Microscopy (Jena, Germany) equipped with a filter eGFP (λex = 489 nm and λem = 509) to detect GFP fluorescence, and an Alexa633 filter (λex = 631 nm and λem = 647 nm) to select for phycocyanin fluorescence.Results and discussion
Engineering of GFPuv and addition of a tyrosine tag (Y-tag)
Various studies reported how crosslinking enzymes recognize residues that are located in flexible, unstructured and exposed regions of proteins. The C-terminal portion of the GFPuv molecule (∼10 amino acids) did not crystallize, thus suggesting flexibility. GFPuv was thus genetically engineered to carry a surface-exposed Y-tag of one or five additional C-terminal tyrosines giving Y1-GFPuv and Y5-GFPuv, respectively (Fig. 1).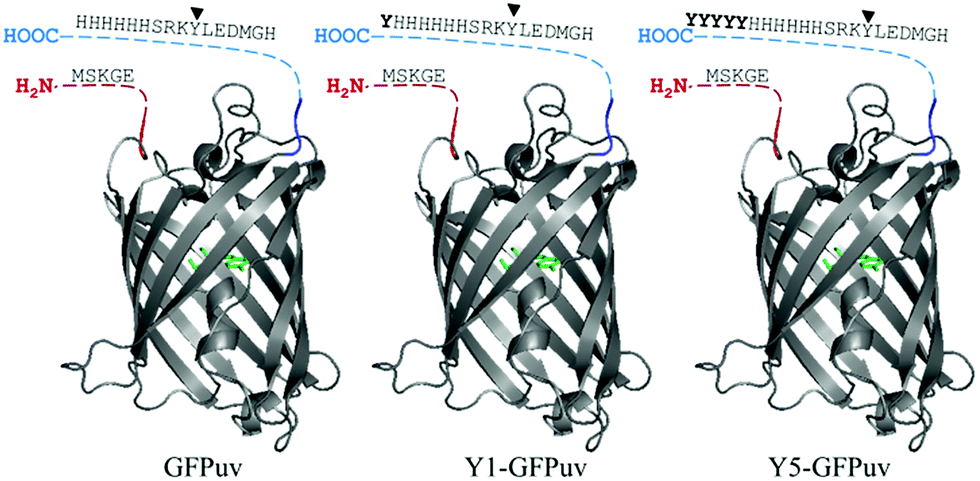 | ||
| Fig. 1 Representation of the original GFPuv molecule (left), engineered to carry a Tyr-(middle) or Tyr5-(left) residue tag. The N- and C-terminal regions not present in the crystallized structure (PDB ID: 1eme, the fluorophore in coloured in green). Tyr237 is indicated by an arrowhead and residues of the Y-tag in bold. | ||
The accessibility of the Y-tag to tyrosinase was assessed with crosslinking experiments in solution. Differently from GFPuv, incubation of Y1- and Y5-GFPuv with tyrosinase led to covalent polymerization with formation of dimeric forms and covalent high molecular weight (HMW) products, e.g. corresponding to multimeric forms with masses above 100 kDa, e.g. corresponding to ∼77 kDa trimers, ∼130 kDa tetramers, and approx. ∼176 kDa likely hexamers (Fig. 2 and S3†). Y-tagged GFPuv variants were efficiently crosslinked even at the lowest tyrosinase concentration tested, e.g. 0.05 mg ml−1, thus, suggesting that the tyrosine residues of the Y-tag were recognized and oxidized by tyrosinase with a higher efficiency than any tyrosine residue from GFPuv.
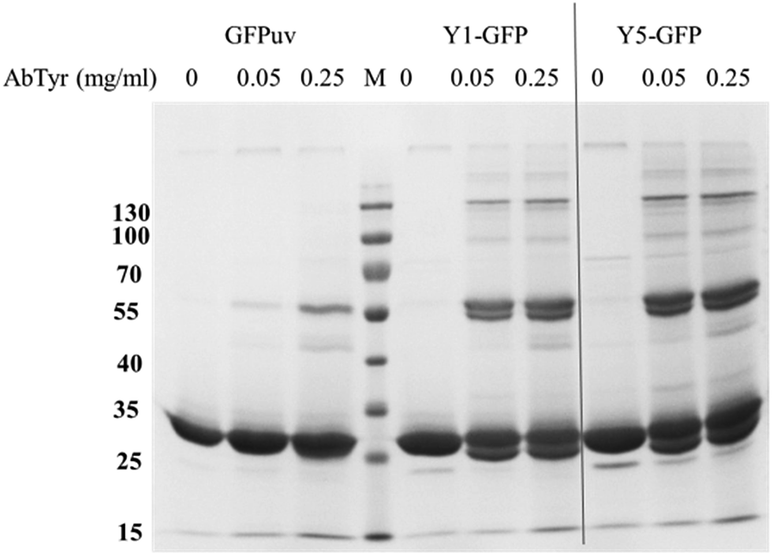 | ||
| Fig. 2 Crosslinking of GFPuv, Y1-GFPuv, and Y5-GFPuv (0.5 mg ml−1) incubated with in the presence of 0, 50, or 250 μg ml−1 tyrosinase for 3 h at 22 °C. Molecular weight markers (lane M) are reported in kDa on the left. | ||
Crosslinking of GFPuv variants with tyrosinase in solution
In order to establish the optimal crosslinking conditions the crosslinking of GFPuv variants was tested using different tyrosinase concentrations and monitored over time.Fig. 2 shows that in the presence of tyrosinase concentrations a covalent GFPuv dimer was also produced, indicating that one tyrosine residue from the native GFPuv molecule was accessible to tyrosinase. The GFPuv molecule carries 11 Tyr residues, including Tyr66 that is involved in the fluorophore formation. A first analysis of the three-dimensional structure of GFPuv (PDB ID: 1eme) identified Tyr39, located in a flexible region between β-strand 2 and 3, and Tyr237, located in the C-terminal region and not present in the crystallized structure (Fig. 1), as possible crosslinking sites. GFPuv variants carrying either Y39F or Y237F substitutions, or a combination thereof, were thus cloned, expressed, purified, and tested for crosslinking by tyrosinase. As crosslinking of GFPuv was observed to larger extent at high tyrosinase concentrations, the influence of different tyrosinase concentrations was tested. The single amino acid substitution Y39F did not affect the crosslinking of GFPuv by tyrosinase. On the contrary, variants GFPuv Y237F and the double mutant GFPuv Y39F-Y237F were still present in the monomeric form after 3 h incubation at any tested tyrosinase concentration (Fig. S3†). Crosslinking proceeded for GFPuv and, at a higher extent, for the Y-tagged forms, e.g. at 0.05 mg ml−1 tyrosinase concentration 37% of Y-tagged GFPuv were in a crosslinked form as compared to 23% of the untagged form carrying Tyr237 (Fig. 3).
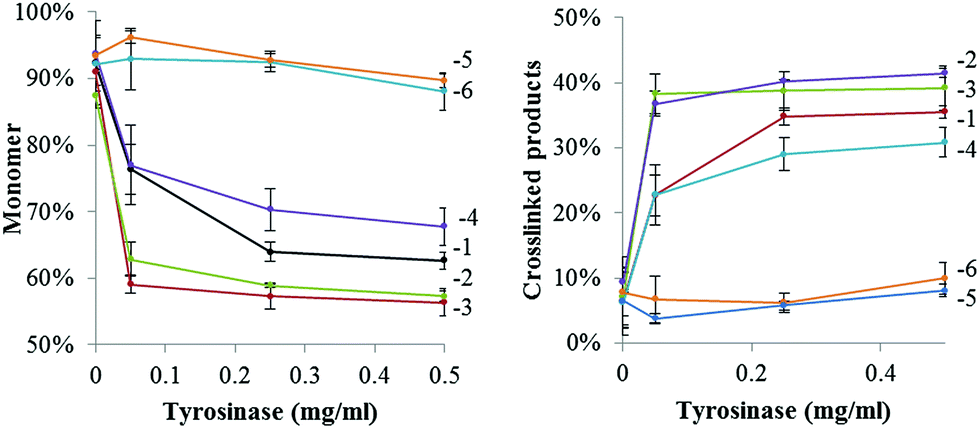 | ||
| Fig. 3 Presence of monomeric (left) and crosslinked (right) forms of GFPuv variants at a 1 mg ml−1 concentration after incubation in the presence of different tyrosinase concentrations for 3 h at 22 °C. Values were obtained by densitometric analyses of protein bands in SDS PAGEs and normalized to the total protein content of each sample. Legend: GFPuv (1), Y1-GFPuv (2), Y5-GFPuv (3), GFPuv Y39F (4), GFPuv Y237F (5), GFPuv Y39F-Y237F (6). Original SDS PAGEs are in Fig. S3.† | ||
We thus concluded that Tyr237 was the residue recognized by tyrosinase and promoted the formation of the covalent dimer of GFPuv. The GFPuv Y237F variant was used as negative control in the later studies.
In order to monitor the progression of the crosslinking reaction, a lower tyrosinase concentration of 0.025 mg ml−1 with a 1 mg ml−1 GFPuv concentration was used. Under these conditions, the GFPuv monomer was converted into the covalent dimeric form but only after long reaction time, e.g. after 24 h. Both the Y-tagged monomeric forms of GFPuv were rapidly crosslinked into covalent dimers and into multimers, e.g. 81% and 79% of Y1-GFP and Y5-GFP, respectively, were left in the monomeric form after 30 min of reaction, whereas 98% of the GFPuv monomer was still present (Fig. 4 and S4†).
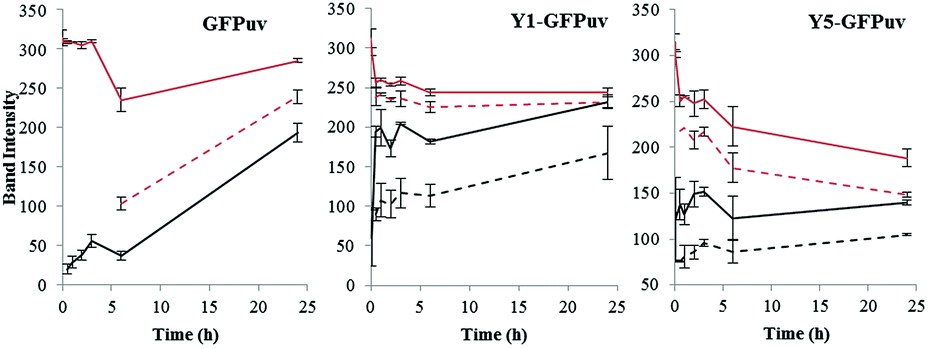 | ||
| Fig. 4 Time course monitoring of the HMW and LMW monomers and dimers produced by crosslinking via tyrosinase of GFPuv (left), Y1-GFPuv (middle), and Y5-GFPuv (right). The intensity of the proteins bands corresponding to monomers (red) and dimers (black), and the corresponding LMW forms (dashed lines) are reported. The original SDS PAGEs are in Fig. S4.† | ||
As can be seen already in Fig. 2, a second dimeric form migrating at a lower molecular weight is produced. In addition, a monomeric form migrating with a lower molecular weight (LMW) was also produced (Fig. S4†). By monitoring the presence of monomeric and dimeric forms, and their LMW counterparts over time (Fig. 4), we established that the monomeric LMW form was not a dead-end product and that it was further crosslinked by tyrosinase, e.g. the intensity of the corresponding band decreased while the one corresponding to the dimer increases over time. The time course experiment revealed the formation of LMW monomers and dimers also from the untagged GFPuv molecule. These LMW forms might be due to intramolecular crosslinking and fragmentation cannot be excluded.21 The crosslinking of GFPuv proceeded slower than that of the Y-tagged forms; no LMW dimer was formed but the LMW monomer was detected after 6 hours. Based on densitometric analyses, the major product of crosslinking of GFPuv was the low molecular weight (LMW) monomer, followed by the dimer and, at a lower rate by the LMW dimer (Fig. 4 and S4†).
SDS PAGE analyses provided only qualitative results. Thus, a quantitative approach for the evaluation of the efficiency of tyrosinase crosslinking on the different variants was performed. By comparing the UV-Vis spectrum of a sample taken from the reaction mixture before and after crosslinking, a major change of absorbance was detected at 290 nm and a smaller one at 475 nm (Fig. S5†). These two wavelengths were then used to monitor the progression of the crosslinking reaction. As seen in Table 1, the presence of a Y-tag resulted in a ∼10-fold higher change of absorbance at 290 nm compared with the untagged version GFPuv. No changes in absorbance were seen with the GFPuv Y237F variant.
| Substrate | Tyrosinase, pH 6.8 | ||
|---|---|---|---|
| ΔA290/min | ΔA475/min | ΔA420/min | |
| L-Tyr | 17.86 | 7.712 | — |
| ABTS | — | — | 3.513 |
| GFPuv | 0.047 | 0.200 | — |
| Y1-GFPuv | 0.542 | 0.091 | — |
| Y5-GFPuv | 0.450 | 0.083 | — |
| GFPuv Y237F | 0.000 | 0.000 | — |
Absorbance changes around 290 nm confirm alteration of the environment of aromatic residues of the protein. A similar change was observed after UV-irradiation of insulin molecules, and attributed to conversion of tyrosine residues to di-tyrosine forms22 although tyrosinase reaction chemistry is different in this case. Similar changes have been ascribed to the formation of thiol–catechol linkages.23 We thus conclude that the location of the tyrosine residue involved in crosslinking affects the efficiency of the crosslinking process, as the terminal Y-tag led to crosslinking with tyrosinase with a higher efficiency than the native residue Tyr237 that is located in a distance of nine residues to the C-terminus, and this was especially evident in batches where low tyrosinase concentrations were applied.
Functionalization of microparticles with tyrosinase
We then proceeded to investigate the tyrosinase-catalyzed immobilization of the fluorescent proteins to amino modified polystyrene microparticles and asked how the amount of catalyst (tyrosinase) and of substrate protein (GFPuv variant) determined the optimal conditions for the immobilization.A tyrosinase concentration of 0.05 mg ml−1 was sufficient to achieve maximum microparticle fluorescence, and no increase was observed with higher tyrosinase amounts (Fig. 5). This concentration of tyrosinase was also used in the following experiments. Since the tyrosinase molecule itself presents surface-exposed amino groups, we then assessed whether tyrosinase molecules were immobilized on the microparticles. Measurement of the tyrosinase activity of the functionalized microparticles showed less than 3% of the initial activity, regardless of the initially applied enzyme concentration (Fig. S6†).
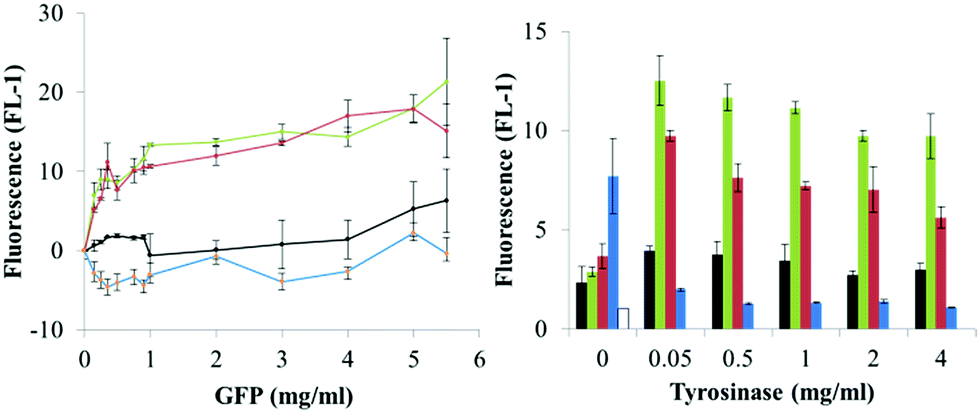 | ||
| Fig. 5 Dependence of microparticles' fluorescence on protein (GFPuv variant) and catalyst (tyrosinase) concentration. Proteins are indicated as GFPuv (1, black), Y1-GFPuv (2, green), Y5-GFPuv (3, red), GFPuv Y237F (4, blue) and untreated microparticles as empty bar. Corresponding microscopy images are in Fig. S8.† | ||
As compared to microparticles incubated with only the GFPuv variants, the additional treatment with tyrosinase led to the production of microparticles with a significantly higher green fluorescence, but only when Y1-GFP and Y5-GFP were used, e.g. a fluorescence of ∼22 versus ∼4 was obtained in the presence and absence of tyrosinase, respectively, using 5 mg ml−1 of Y-tagged GFPuv. Remarkably, microparticles treated with Y-tagged GFPuv and tyrosinase had a fluorescence ∼10-fold higher that the ones treated with GFPuv and tyrosinase (Fig. 5). The strongest development in particle fluorescence was detected with a concentration of Y-tagged GFPuv between 0.15 and 1 mg ml−1. In this case a less significant dependence on the GFPuv concentration was found. Considering the amount of GFP molecules used in the immobilization process and the final fluorescence of the microparticles, Y-tagged GFPuv variants could be immobilized more efficiently than the untagged variants, especially at the lowest tyrosinase concentration tested, (0.15 mg ml−1) (Fig. S7†). Similarly to what is reported for protein–protein crosslinking, these results indicated that the number of tyrosine residues presented in the Y-tag did not affect the extent of immobilization with tyrosinase. The location of such residues seems to have more impact on successful immobilization.
Multi-functionalization of microparticles with Y1-GFPuv and HisCPC with tyrosinase
We previously reported the crosslinking and immobilization of the fluorescent protein HisCPC with tyrosinase. Interested in the production of multi-functional microparticles using crosslinking enzymes, we thus proceeded to test the co-immobilization of Y1-GFP and HisCPC with tyrosinase. The microparticles were analysed by flow cytometry and imaged by fluorescence microscopy. Microparticles treated with tyrosinase and Y1-GFP acquired a ∼6 fold higher green fluorescence than equivalent beads incubated in the absence of tyrosinase, also when incubated in the presence of both Y1-GFP and HisCPC (Fig. S9†). Imaged by microscopy a clear co-localization of GFP- and HisCPC-specific fluorescence was visualized on the microparticles (Fig. 6 and S9†). Imaged by CLSM, a well-defined, homogenous, and smooth thin layer with an apparent thickness of 0.5 μm and showing both GFP and CPC fluorescence was seen. This indicates that both proteins co-localized at the surface. Crosslinking experiments in solution and the analysis of the immobilization in solution at the endpoint of the reaction showed that no Y1-GFP–HisCPC ∼45 kDa hetero-conjugates were produced by tyrosinase, indicating the independent immobilization of the two fluorescent proteins (Fig. S10†). A likely explanation might be provided by the electrostatic repulsion occurring at pH 6.8 between the GFPuv molecule with a pI of ∼5 and HisCPC with a pI of 4.6, which were both negatively charged. The formation of hetero-multimers might thus be possible in solution with proteins that are differently charged under the reaction conditions.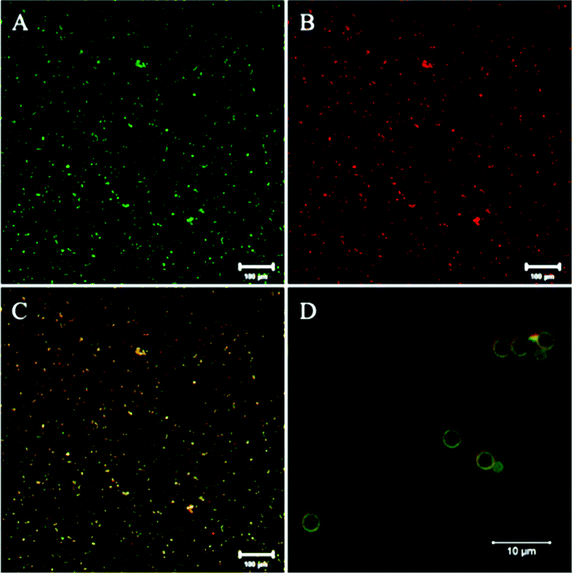 | ||
| Fig. 6 CLSM imaging of microparticles functionalized with both Y1-GFP and HisCPC using tyrosinase. Microparticles were imaged for GFP fluorescence (green, A) and CPC fluorescence (red, B) at a 10× (A–C) and 63× (D) magnification. Single fluorescence signals (A and B) and their combination (C and D) are shown (enhanced contrast for clarity). | ||
Conclusions
The combined use of a crosslinking enzyme such as tyrosinase and the possibility of ad hoc genetically engineering proteins allowed materials surface functionalization with changed optical properties. The immobilization process could take place in aqueous solutions under mild reaction conditions, and using catalytic amounts of tyrosinase enzyme. This enzymatic approach for the production of particles and surfaces carrying multiple proteins proceeds efficiently especially at low GFPuv concentration. This is particularly advantageous when limited amounts of protein are available for immobilization, e.g. due to difficult recombinant production or elevated costs of the raw material. The reactivity of tyrosinase with tyrosine residues from a protein can lead to different processes taking place in solution (Fig. 7). Our work proposes an enzyme-based immobilization process under mild conditions that is advantageous when the close proximity of different proteins on a carrier is necessary such as on microparticles, e.g. in the case of multi-enzymatic processes where the reaction intermediates need to efficiently transfer through the steps of an enzymatic cascade reaction.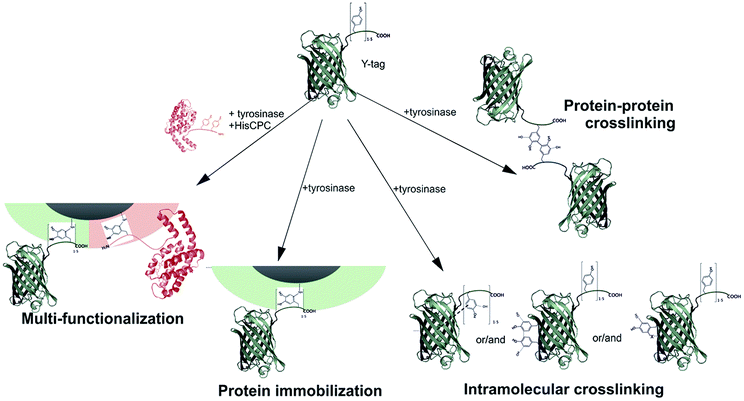 | ||
| Fig. 7 Schematic representations of the possible reactions taking place upon incubation of Y-tagged GFPuv with tyrosinase and leading to protein–protein crosslinking, the production of an intramolecular crosslinked molecules, and protein immobilization to the surface. For ease of visualization, only Tyr–Tyr crosslinks are shown. However, Tyr–Lys/His/Cys are also possible. | ||
Acknowledgements
We are grateful to Arie Bruinink and Gökce Yazgan for the assistance with CLSM. GF was funded by the TYROMAT project (COFUND action between Empa Postdocs programme and FP7: People) and by PHOTOTEC, a project funded by the State Secretariat for Education, Research and Innovation (SERI) within the COST action TD1102.Notes and references
- P. Jochems, Y. Satyawali, L. Diels and W. Dejonghe, Green Chem., 2011, 13(7), 1609 RSC.
- T. Heck, G. Faccio, M. Richter and L. Thöny-Meyer, Appl. Microbiol. Biotechnol., 2013, 97, 461 CrossRef CAS PubMed.
- J. Buchert, D. Ercili Cura, H. Ma, C. Gasparetti, E. Monogioudi, G. Faccio, M. Mattinen, H. Boer, R. Partanen, E. Selinheimo, R. Lantto and K. Kruus, Annu. Rev. Food Sci. Technol., 2010, 1, 113 CrossRef CAS PubMed.
- H. Leutbecher, S. Hajdok, C. Braunberger, M. Neumann, S. Mika, J. Conrad and U. Beifuss, Green Chem., 2009, 11, 676 RSC.
- R. Jin, B. Lou and C. Lin, Polym. Int., 2013, 62, 353 CrossRef CAS.
- C. R. Thalmann and T. Lotzbeyer, Eur. Food Res. Technol., 2002, 214, 276 CrossRef CAS PubMed.
- B. Zeeb, L. Fischer and J. Weiss, Food Funct., 2014, 5, 198 CAS.
- S. Isaschar-Ovdat, M. Rosenberg, U. Lesmes and A. Fishman, Food Hydrocolloids, 2014, 43, 493 CrossRef PubMed.
- W. T. Ismaya, H. J. Rozeboom, A. Weijn, J. J. Mes, F. Fusetti, H. J. Wichers and B. W. Dijkstra, Biochemistry, 2011, 50(24), 5477 CrossRef CAS PubMed.
- E. Jacob and R. Unger, Bioinformatics, 2007, 23, e225 CrossRef CAS PubMed.
- A. T. Lewandowski, D. A. Small, T. H. Chen, G. F. Payne and W. E. Bentley, Biotechnol. Bioeng., 2006, 93, 1207 CrossRef CAS PubMed.
- A. T. Lewandowski, H. Yi, X. Luo, G. F. Payne, R. Ghodssi, G. W. Rubloff and W. E. Bentley, Biotechnol. Bioeng., 2008, 99, 499 CrossRef CAS PubMed.
- G. Faccio, M. M. Kämpf, C. Piatti, L. Thöny-Meyer and M. Richter, Sci. Rep., 2014, 4, 5370 Search PubMed.
- F. Thielbeer, S. V. Chankeshwara and M. Bradley, Biomacromolecules, 2011, 12, 4386 CrossRef CAS PubMed.
- A. Sassolas, L. J. Blum and B. Leca-Bouvier, Biotechnol. Adv., 2012, 30, 489 CrossRef CAS PubMed.
- V. Sokolova and M. Epple, Nanoscale, 2011, 3, 1957 RSC.
- A. Crameri, E. A. Whitehorn, E. Tate and W. P. C. Stemmer, Nat. Biotechnol., 1996, 14, 315 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671 CrossRef CAS.
- T. Heck, P. Pham, F. Hammes, L. Thöny-Meyer and M. Richter, Bioconjugate Chem., 2014, 25, 1492 CrossRef CAS PubMed.
- D. G. Gibson, L. Young, R. Chuang, J. C. Venter, C. A. Hutchison and H. O. Smith, Nat. Methods, 2009, 6, 343 CrossRef CAS PubMed.
- M. J. C. Long and L. Hedstrom, ChemBioChem, 2012, 13, 1818 CrossRef CAS PubMed.
- M. Correia, M. Neves-Petersen, P. B. Jeppesen, S. Gregersen and S. B. Petersen, PLoS One, 2012, 7, e50733 CAS.
- Y. Lee, H. J. Chung, S. Yeo, C. Ahn, H. Lee, P. B. Messersmith and T. G. Park, Soft Matter, 2010, 6, 977 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra00669d |
| This journal is © The Royal Society of Chemistry 2015 |