
Bead beating-based continuous flow cell lysis in a microfluidic device
A. Berasaluce*ab,
L. Matthysb,
J. Mujikab,
M. Antoñana-Díezab,
A. Valeroa and
M. Agirregabiriaab
aCIC-Microgune, Polo Garaia, Goiru 9, 20500 Arrasate, Spain
bIK4-Ikerlan, Polo Garaia, Goiru 9, 20500 Arrasate, Spain. E-mail: aberasaluce@ikerlan.es
First published on 20th February 2015
Abstract
This paper describes a bead beating-based miniaturized cell lysis device that works in continuous flow allowing the analysis of large volumes of samples without previous treatment. A permanent magnet along with zirconium/silica beads were placed inside a lysis chamber fabricated with cyclo-olefin polymer (COP) by a fast prototyping technique, and the actuation of an external magnetic field caused the motion of the beads within the chamber. Characterisation of the lysis process was carried out using Staphylococcus epidermidis as the target cell and showed that both small bead size and large volume, along with the presence of Tween 20 and low flow rate, influenced significantly the device performance. Taking into account the compromise between time consumption and efficiency, 60 μL min−1 lysis flow rate was chosen as optimum yielding 43% lysis efficiency relative to off chip bead beating. Compatibility with injection moulding manufacturing techniques and capability of working in continuous flow make this device a potential DNA extraction method suitable for lab-on-a-chip applications.
1. Introduction
Early detection of microorganisms is required in many health-related fields to avoid possible infections.1 The traditional gold standard methods for bacteria identification and counting in microbiology have relied on culture-based diagnostics,2,3 which is a time consuming method. On the other hand, deoxyribonucleic acid (DNA) detection kits have appeared as commercial alternatives.4–6 Nevertheless, these comprise many sample manipulation steps and need a qualified worker, deriving in potential cross contamination risks. The solution to overcome these limitations arises in integrating different analysis steps (i.e. sample preparation, DNA amplification and detection) into an automated lab-on-a chip (LoC) device.The accepted method to amplify and detect DNA molecules is real-time polymerase chain reaction (qPCR), a sensitive technique suitable for microfluidic integration.7,8 Its performance depends on previous DNA extraction from target cells, which in turn is cell wall disruption dependent, especially working with microorganisms that show resistivity to lyse such as Gram positive bacteria and fungi.9,10
Many works have reported microfluidic integrated DNA extraction methods. They can be roughly classified into two main groups: chemical methods and physical methods.
Chemical methods release cellular DNA by solubilising membrane lipids and proteins using chaotropic agents such as guanidine thiocyanate11,12 and biological enzymes.13 They are easy to implement as they do not need any additional equipment. However, a prior mixing step is needed and this can be difficult to achieve on chip as the flow regime is laminar. Furthermore, the employed chemicals can inhibit subsequent downstream reactions, thus a thorough cleaning step is necessary.14
Generally, physical methods require additional hardware components that increase complexity for microsystem integration. Conversely they present many advantages, since they do not leave residual substances, are faster and more efficient. Several physical lysis methods have been reported based on thermal treatment,15 sonolysis,16 electroporation,17 laser induced cell wall disruption18 and mechanical lysis.19 Among them, mechanical lysis based on bead beating is the commonly used method working with hard to lyse samples because of its effectiveness and reproducibility.20 Considering these advantages, Claremont BioSolutions has worked on different devices capable of lysing Gram positive bacteria (B. Subtilis and M. Bovis) achieving efficiencies as high as benchtop products within 2 minutes.21,22 However, such products also present some drawbacks; OmmiLyse Bead Blender is an off-chip device and Micro Bead-Beater requires tubes and pertinent connections for sample transport, hindering integration into a monolithic sample to answer system.
Some authors along the line have successfully implemented the bead beating strategy into microfluidic systems. Siegrist et al.23 adapted potential advantages of centrifugation into a microfluidic compact-disc labcard. The rotation of the chip actuated a permanent magnet located in the lysis chamber, causing intense mixing and subsequent collision between beads and cells. Nonetheless, sample volume was limited by the chamber volume (70 μL). Hwang et al.24 concentrated high volume of bacteria samples employing functionalized glass beads packed in a chamber and achieved lysis by pneumatic vibration of an elastomeric membrane of PDMS. Although this material is a good candidate for fabricating microfluidic prototypes by moulding, it also presents several disadvantages: (i) its aging affects the mechanical properties, (ii) it can release undesired contaminants to the sample that can be harmful for biological reactions if bad cross-linking process occurs and, (iii) it is not the best material for mass production. From a commercial point of view, polymers such as cyclic olefin copolymer (COC) or COP are the most suitable materials due to their compatibility with injection moulding fabrication techniques as well as their biocompatibility.
In this work we present a microfluidic device made of COP that combines a magnetic stirrer and bead beating features for cell wall disruption of hard to lyse microorganisms. The system is thought to be used in applications which require large sample volumes, such as nasal, oral and water bacteria analysis. In order to minimize time consumption, our system works in continuous flow performing lysis at flow rates ranging from 30 μL min−1 to 180 μL min−1. Its capability to process large volumes avoids previous off-chip treatment and the use of both chemical reagents and heat which can inhibit downstream PCR reactions and/or protein analysis.
2. Materials and methods
2.1. Device fabrication
The cell lysis device was fabricated by a fast prototyping technique resulting in a monolithic microfluidic structure.25 It consisted of self-alignment folding, stacking and bonding of 100 μm thick COP foils (ZEON chemicals) previously structured by a cutting plotter (GRAPHTEC FC8000-601). A schematic layout and a digital picture of the chip are depicted in Fig. 1. Zirconia/silica beads (BioSpec) and a NdFeB stirring permanent magnet (Supermagnete) (1.3 × 1.3 × 6 mm) with radial magnetization were accommodated in a lysis chamber of 115 μL and sealed with pressure sensitive adhesive (Progene from Ultident). The lysis chamber connected the sample inlet and outlet via 400 μm high channels that became narrower, down to 100 μm just before reaching the chamber. In this way, chamber outlet and inlet showed low fluidic resistance and worked as bead weir at the same time, since the smallest beads were larger than 100 μm in diameter. Luer connectors made by stereolithography were used for the fluidic connections to the sample inlet and outlet.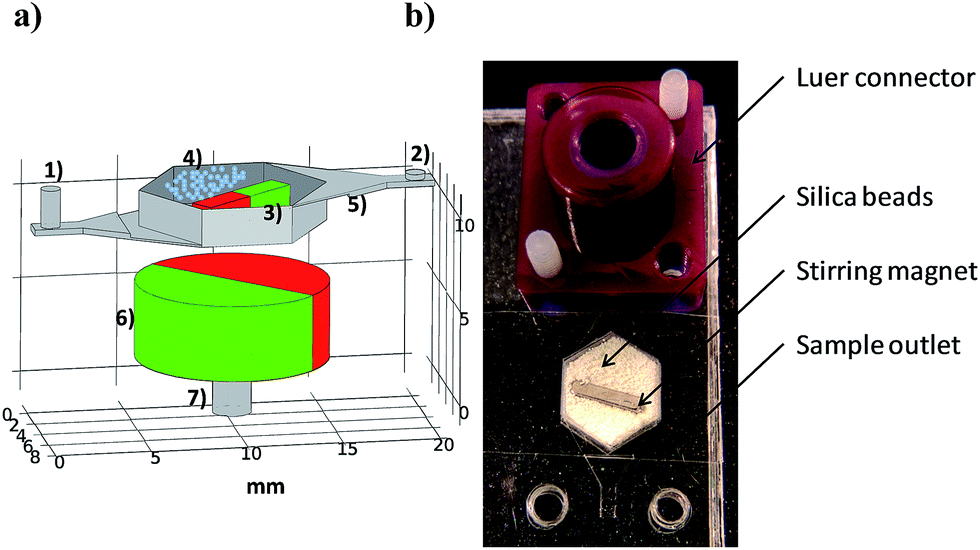 | ||
| Fig. 1 Bead beating device: (a) schematic illustration with all the components: (1) inlet, (2) outlet, (3) stirring magnet, (4) zirconia/silica beads, (5) bead weir, (6) rotating magnet and (7) electric motor coupling. (b) Digital image of the system ready to lyse. | ||
2.2. Experimental set-up
A permanent magnet (12 mm in diameter and 4 mm long cylindrical body) with radial magnetization coupled to an electrical motor (Maxon A-max, 12 mm in diameter and 21 mm long) was located in the vertical axis of the chamber (see Fig. 1 (6) and (7)). The distance between the rotating magnet and the bottom of the chip was 4 mm.The chamber inlet was connected to a syringe containing the sample via a Luer connector, whereas the outlet was connected to an empty syringe. The plunger of the sample syringe was computer controlled displaced at a determinate flow rate by a mechanical pusher. While the sample flowed through the system, the magnet was rotating due to the magnetic field rotation caused by the permanent magnet coupled to the electrical motor. Hence, a bead beating-based bacteria lysis device in continuous flow was achieved. The lysate product was recovered in the outlet syringe.
2.3. Cell strain and culture
Gram positive Staphylococcus epidermidis (ATCC# 12228TM) was used for the verification and optimization of the system. Strains were grown for 5 hours under aerobic conditions at 37 °C using trypticase in soy agar sheep blood plates. Grown colonies were recovered with a sterile Digralsky spreader and resuspended in 1 mL of phosphate-buffered saline (PBS). Resuspended cells were twice harvested by centrifugation at 5000 rpm for two minutes and washed with PBS in order to eliminate supernatant DNA. Bacteria concentration was calculated from plate counting by plating 10 fold dilutions, and required dilutions were done with 20% glycerol–PBS to a final concentration of 107 and 106 CFU mL−1. These solutions were divided into 100 μL aliquots and stored at −80 °C.2.4. DNA extraction
Frozen bacteria aliquots were thawed at room temperature and diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 in PBS. Syringes were filled with 450 μL of sample and connected to the Luer connector of the device. The plunger pusher was actuated and when the suspension wetted the chamber, the electric motor was switched on. The lysate recovered in the outlet syringe was stored in the fridge until its analysis in the thermocycler.
:10 in PBS. Syringes were filled with 450 μL of sample and connected to the Luer connector of the device. The plunger pusher was actuated and when the suspension wetted the chamber, the electric motor was switched on. The lysate recovered in the outlet syringe was stored in the fridge until its analysis in the thermocycler.
2.5. Real time PCR amplification
Real-time polymerase chain reaction was performed on a Biorad CFX96 Touch for DNA detection by targeting a nucleic acid metabolism related gene (gmk) of S. epidermidis.26 The reaction mixture was composed of 10 μL Premix Ex TaqTM (Takara), 2 μL of primers and probe with a final concentration of 500 nM and 250 nM, respectively 4 μL of nuclease free water and 4 μL sample, yielding an amplicon of 93 bp. Primers and probe were purchased from IDT (Table 1). Cycling conditions were: 90 s at 95 °C (initialization), 40 cycles of 15 s at 95 °C (denaturalization) and 45 s at 59 °C (annealing and elongation).| Microorganism | Forward 5′–3′ | Reverse 5′–3′ | Probe 5′–3′ |
|---|---|---|---|
| Staphylococcus epidermidis26 | CAACAAGACGTTCTTTCAAGTCATCT | AAGGTGCTAAGCAAGTAAGAAAGAAATT | /56-FAM/ATGCGTTGT/ZEN/TCATA TTTTTAGCGCCTCCA/3IABkFQ/ |
| Methicillin resistant Staphylococcus aureus27 | CATTGATCGCAACGTTCAATTT | TGGTCTTTCTGCATTCCTGGA | /5TET/TGGAAGTTA/ZEN/GATT GGGATCATAGCGTCAT/3IABkFQ/ |
| Streptococcus uberis28 | AGAGGAATTCATCATGTTTTAACA | AATTGTAGAAGAACCATTTGATGT | /56-FAM/AGCGTCTAACAAC TCGGCCTTTG/3IABKFQ/ |
Purified genomic DNA (12228D-5™) was used to build an external standard curve to quantify lysis performance and validate linearity in the operating concentration range. Efficiency of the qPCR was calculated by plotting the cycle threshold versus the logarithmic concentration of the standard DNA in 4 consecutive days, yielding 100 ± 0.1% efficiency. The cycle threshold was set at 100 arbitrary units for all the experiments.
2.6. Lysis efficiency
Lysis efficiency was calculated relative to a well established off-chip lysis method.29 250 μL sample along with 650 mg zirconia/silica beads of 100 mm in diameter were added to 2 mL tubes and vortexed for 2 minutes in triplicate. Bacteria concentration was measured by plate counting before and after bead beating, showing that this off-chip method was capable to lyse >99% of the initial colony forming units. This method was considered 100% efficient.On the other hand, qPCR efficiency resulted close to 100%, which means that in each cycle the DNA concentration was doubled. Therefore, lysis efficiency was calculated by the following equation:

| ΔCt = Ctin chip − Ctoff-chip (bead beating) |
The possible contribution of unlysed bacteria to the lysis efficiency due to the thermocycling heating steps of the qPCR was measured and considered as a control. For this purpose, the Ct values of untreated samples and samples lysed by off-chip bead beating were compared. The ΔCt between both was of 7.7 cycles. Thus, according to the equations shown above, the supernatant DNA and DNA released during the qPCR heating steps represented the 0.5% of the total amount of DNA. Therefore, DNA contribution of the unlysed bacteria was considered negligible.
2.7. Variables optimization
A fractional factorial experimental design (STATISTICA 10) was carried out to identify variables influencing significantly on the lysis process. The selected variables with their major and minor fixed values are summarized in Table 2. A negative control variable was considered for validation purposes, which did not change experimental conditions. Along the variable screening, 16 experiments in replicate were done divided into 4 blocks, one per day, resulting in a 2(7−3) design. This was done to avoid the variability inherent to daily sample preparation process such as sample manipulation, sample aliquoting, etc. Input data of cycle differences between the crossing points (ΔCT) of the initial solution and each lysate were used to calculate ANOVA effect estimates of each variable on lysis performance and built a ΔCT prediction model.| Minor value | Major value | |
|---|---|---|
| a Bead quantity refers to the free volume occupied by beads in the chamber.b The voltage applied to the electric motor. | ||
| Bead diameter | 100 μm | 200 μm |
| Bead quantitya | 40% | 45% |
| Flow rate | 30 μL min−1 | 60 μL min−1 |
| Bacteria conc. | 105 CFU mL−1 | 106 CFU mL−1 |
| Stirrer voltageb | 7 V (5200 rpm) | 9 V (6800 rpm) |
| Tween 20 | Absence | 0.05% |
| Control (neg) | — | — |
One of the chosen variables for the study was the size of the beads. Two different sizes were purchased, catalogued as 100 and 200 μm in diameter by the suppliers. Beads were examined under the microscope (Fig. 2) and their diameter measured by ImageJ software. A statistical population of 55 samples gave a mean diameter value of 194 ± 19 μm for small beads and the mean value for 33 samples of the large beads was 355 ± 49 μm.
 | ||
| Fig. 2 Microscope images of the beads used. Beads are catalogued as 100 μm (a) and 200 μm (b) in diameter. Pictures were taken with 60:1 visual magnification. | ||
3. Results and discussion
3.1. First trials
First attempts were carried out with beads of 200 μm in diameter occupying 35% of the chamber and the electric motor was set at 11 V. The sample was introduced at a constant flow rate of 50 μL min−1. As a result, the qPCR signal of the lysate increased in 4 cycles compared to the initial solution, proving that on-chip continuous flow bead beating was able to release DNA from Gram positive bacteria cells (see Fig. 3). Nevertheless, the sigmoidal curve of the lysate sample reaches lower plateau value and its slope is smaller than the initial solution sample.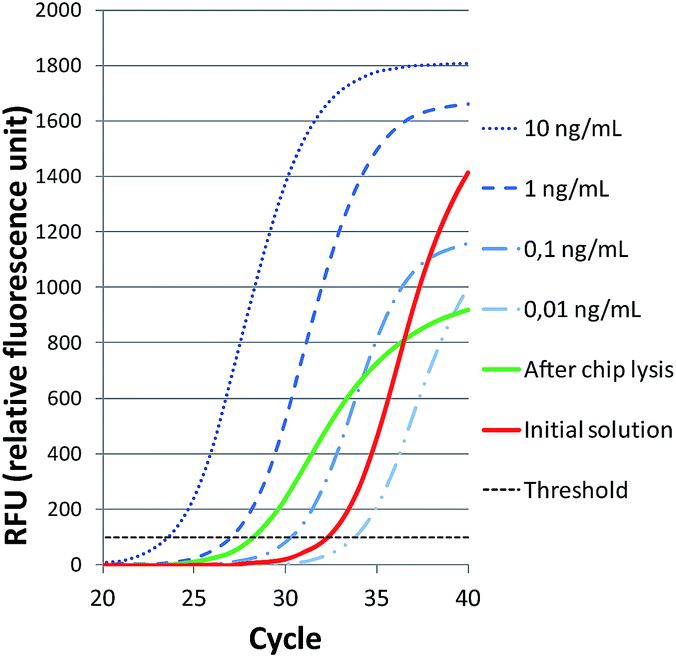 | ||
| Fig. 3 Real-time PCR detection of Staphylococcus epidermidis. Amplification curves of standard DNA 10 fold dilutions used to calculate qPCR efficiency and 105 CFU mL−1 bacteria solution signal before and after on-chip lysis. | ||
To assess possible inhibitions or other undesired effects when lysate solution was introduced into the thermocycler without any purification step, an internal control was added to the reaction mixture. 4 μL of nuclease free water used in the initial reaction mixture was replaced with 1 μL of methicillin resistant Staphylococcus aureus (MRSA) genomic DNA (33591D-5™), 2 μL of primers and probe designed to detect mecA gene of MRSA (Table 1) and 1 μL of nuclease free water. Compatibility between primers and probes used in the duplex reaction was checked by Oligo Analyzer 3.1 software (IDT). The polymerase chain reaction was run under the same thermocycling conditions as described previously.
The study was carried out comparing qPCR results of eight lysis replicates containing the internal control with eight replicates containing just the internal control. The Ct average values of the internal control were similar for samples with lysate and without lysate, 31.3 ± 0.2 and 31.1 ± 0.1, respectively. This means that there are not adverse effects during the logarithmic phase of the amplification process and characterization of the lysis process can be done without any purification of the cellular debris.
3.2. Significant factors affecting cell disruption
Once it was verified that the lysis performance could be measured by qPCR, several parameters had to be optimized in order to achieve maximum efficiency. The experimental design helped to choose the convenient value of variables affecting notably on the lysis process.The r square value of the built model was 0.8, suggesting the model fitted considerably. Variables influence on the lysis process is shown in Table 3. Highlighted in bold are variables affecting significantly (p < 0.05) and the values that improve the efficiency (taken from the Pareto graph, not shown).
| Value | P | |
|---|---|---|
| Blocks | — | <0.01 |
| Bacteria conc. | — | 0.68 |
| Bead size | 100 μm | <0.01 |
| Lysis time | 2 min | <0.01 |
| Bead quantity | 45% | 0.03 |
| Stirrer voltage | — | 0.60 |
| Tween 20 | 0.05% | 0.02 |
| Control (neg) | — | 0.88 |
Results show that both bead size and bead quantity affect notably on efficiency. Small beads size and 45% of chamber filling increases the amount of released DNA, possibly due to the higher probability of collision and shear. Nevertheless, none of these variables are suitable for subsequent optimization. On one hand, smaller beads could go through the barrier increasing the fluidic resistance or even clogging the channels, and on the other hand, larger amount of beads in the lysis chamber hinders the rotation of the magnet. Likewise, usage of Tween 20 improves DNA recovery, since its detergent nature weakens the cell wall.30 However, higher concentration of surfactant brings on bubble formation, which is a problem to avoid in microfluidics. The fourth variable is the flow rate. The lower the flow rate the higher the cell lysis efficiency, but low flow rates can be a drawback in case that a minimum time-to-results is required. Finally, it must be remarked that the partition of the experimental design in 4 blocks has eliminated the intervariability of the experiments.
With the adjustment of all these factors, the cycle difference between the initial solution and lysate has been increased from 4 cycles (see Fig. 3) up to 6.5 cycles.
3.3. Lysis efficiency versus flow rate
Flow rate was the only adjustable significant variable, so that the effect of the flow rate on lysis efficiency was further studied. Three lysis replicates were performed at four different flow rate values (30, 60, 90 and 180 μL min−1), while the other parameters remained unchanged (i.e. 100 μm bead size, 45% bead quantity, 0.05% Tween 20, 106 CFU mL−1 and 7 V).Results are shown in Fig. 4, where it can be seen that the lowest flow rate achieves the highest efficiency (56%). On the contrary, the highest flow rate gives 30% lysis efficiency within three minutes. Summarizing, fast lysis process leads to a poor lysis efficiency while the opposite situation is a time consuming efficient process. Moreover, the higher the flow rate the lower the deviation of the measurements.
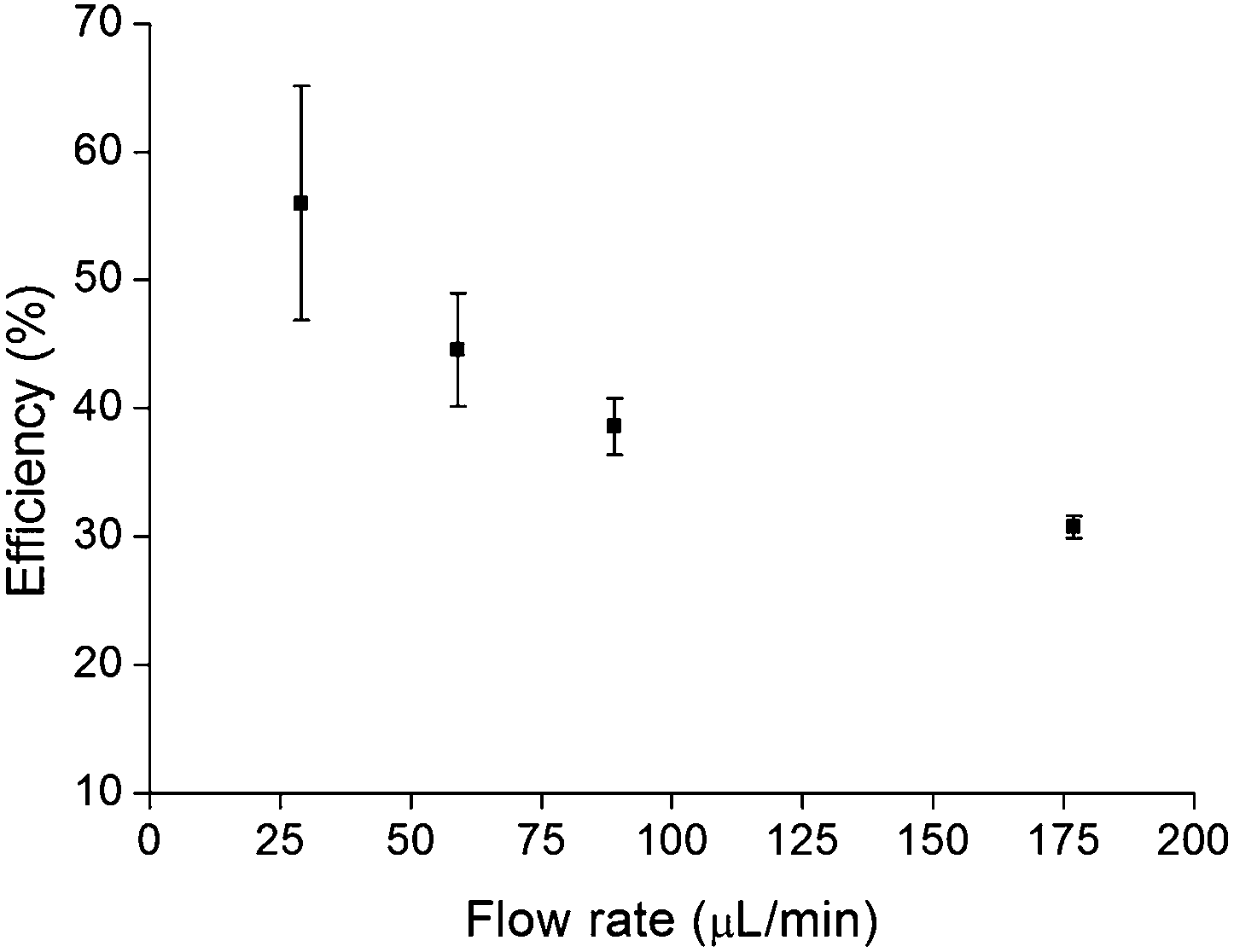 | ||
| Fig. 4 Effect of flow rate on DNA recovery efficiency in relation to off-chip bead beating vortex system. Three replicates were performed at each flow rate. | ||
Considering time to results and efficiency, 60 μL min−1 flow rate was chosen as the optimal flow rate. The whole lysis process takes 8 minutes.
3.4. Reproducibility
In order to evaluate reproducibility, 12 replicates were run and showed high reproducibility. Results gave a 43 ± 3% of cell lysis efficiency in comparison with the off-chip vortex method. In addition, these results are in agreement with those obtained at a flow rate of 60 μL min−1 (see Section 3.3). Despite the deviation approximates to 6%, it is much lower than the deviation that can arise during sampling (swab selection,31 sampling32 and sample transport system selection33) in nasal or oral bacteria analysis processes. So, reproducibility of the method is in accordance with our requirements.3.5. Streptococcus uberis
Our device was validated with another Gram positive bacterium using Streptococcus uberis (ATCC#27958) as target. Bacteria samples with concentrations of 3 × 108, 3 × 107 and 3 × 106 CFU mL−1 were prepared as described previously in Section 2.3. Lysis efficiency was determined comparing the off-chip method (duplicate) with the on-chip (triplicate) by means of qPCR. Thermocycling conditions and protocol were the same as with S. epidermidis. The primers and probe were specific for S. uberis (Table 1). Calibration curves made with standard DNA resulted in 104% efficiency ensuring linearity within the working DNA concentration range.Lysis efficiencies were 39 ± 5, 36 ± 2 and 41 ± 6 which correspond to 3 × 106, 3 × 107 and 3 × 108 CFU mL−1 respectively as shown in Fig. 5. Firstly, no trend is observed among different initial bacteria concentration, corroborating the fact that lysis efficiency is not bacterial concentration dependant as concluded in the experimental design. The existing variability can be a consequence of using a single quencher probe, which increases background signal and subsequently reduces sensibility and precision of the detection.34 And secondly, method performance is close to that observed with S. epidermidis.
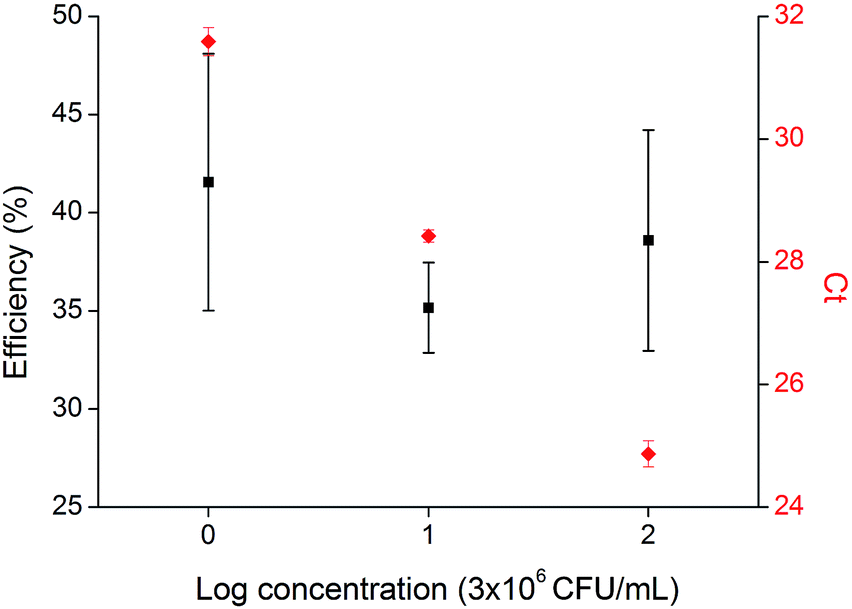 | ||
| Fig. 5 Lysis efficiency relative to off-chip method and qPCR cycle threshold for 3 × 108, 3 × 107 and 3 × 106 CFU mL−1 Streptococcus uberis. Three replicates were carried out for each cell number. | ||
4. Conclusions
Miniaturization of bacterial detection in a lab-on-a-chip device has been a challenging issue during the past few years. In this context, we have developed an efficient cell lysis technique based on bead beating capable of extracting DNA from Gram positive bacteria without leaving any residual molecules that can inhibit subsequent amplification reactions. It works in continuous flow and lyse large sample volumes. The system was characterized using S. epidermidis as target and its performance was also proved with S. uberis.Although it was fabricated by a rapid prototyping technique, its design is fully compatible with mass production by injection moulding. The easy fabrication process and the relative small volume it occupies make this system suitable for future incorporation into an automated microorganism detection device.
Acknowledgements
This work was supported by Gobierno Vasco, DEPARTAMENTO DE DESARROLLO ECONÓMICO Y COMPETITIVIDAD under the program EMAITEK and Diputación Foral de Gipuzkoa under the program Red Guipuzcona de Ciencia, Tecnología e Innovación.Notes and references
- L. G. Bode, J. A. Kluytmans, H. F. Wertheim, D. Bogaers, C. M. Vandenbroucke-Grauls, R. Roosendall, A. Troelstra, A. T. Box, A. Voss, I. van der Tweel, A. van Belkum, H. A. Verbrugh and M. C. Vos, N. Engl. J. Med., 2010, 362, 9–17 CrossRef CAS PubMed.
- B. Lungu, W. Douglas, R. Berghaus and C. Hofacre, J. Food Prot., 2011, 75, 743–747 CrossRef PubMed.
- J. Burns and J. Rolain, J. Cystic Fibrosis, 2013, 13, 1–9 CrossRef PubMed.
- E. L. Palavecino, Methods Mol. Biol., 2014, 1085, 71–83 Search PubMed.
- S. Boyle-Vavra and R. S. Daum, J. Clin. Microbiol., 2010, 48, 4546–4551 CrossRef PubMed.
- G. Andriesse, M. van Rijen, D. Bogaers, A. M. Bergmans and J. A. Kluytmans, Clin Microbiol Infect, 2009, 10, 1223–1226 Search PubMed.
- E. Oblath, W. Henley, J. Alarie and J. M. Ramsey, Lab Chip, 2013, 13, 1325–1332 RSC.
- C. Zhang and D. Xing, Nucleic Acids Res., 2007, 35, 4223–4237 CrossRef CAS PubMed.
- T. Gosiewski, L. Szala, A. Pietrzyk, M. Brychczy-Wloch, P. Hechzko and M. Bulanda, Curr. Microbiol., 2014, 68, 149–155 CrossRef CAS PubMed.
- M. Mahalanabis, H. Al-Muayad, M. D. Kulinski, D. Altman and C. M. Klapperich, Lab Chip, 2009, 9, 2811–2817 RSC.
- L. Heirstraeten, P. Spang, C. Schwind, K. Drese, M. Ritzi-Lehnert, B. Nieto, M. Camps, B. Landgraf, F. Guasch, A. N. Corbera, J. Samitier, H. Goossens, S. Malhotra-Kumar and T. Roeser, Lab Chip, 2014, 14, 1519–1526 RSC.
- A. F. Sauer-Budge, P. Mirer, A. Chatterjee, C. Klapperich, D. Chargin and A. Sharon, Lab Chip, 2009, 9, 2803–2810 RSC.
- O. Salazar and J. A. Asenjo, Biotechnol. Lett., 2007, 29, 985–994 CrossRef CAS PubMed.
- L. Nang, Z. Jiang and X. Wei, Lab Chip, 2014, 14, 1060–1073 RSC.
- C. Wang, K. Lien, J. Wu and G. Lee, Lab Chip, 2011, 11, 1521–1531 RSC.
- T. Tandiono, D. Ow, L. Driessen, L. Chin, E. Klaseboer, A. Choo, S. Ohl and C. Ohl, Lab Chip, 2011, 12, 780–786 RSC.
- A. Talebpour, R. Maaskant, A. Khine and T. Alavie, PLoS One, 2014, 9, e102707 Search PubMed.
- J. Lee, K. Cheong, N. Huh, S. Kim, J. Choi and C. Ko, Lab Chip, 2005, 6, 886–895 RSC.
- J. Kim, J. Woo Hong, D. Pyo Kim, J. H. Shin and I. Park, Lab Chip, 2012, 12, 2914–2921 RSC.
- M. Hhnadel, L. Felden, D. Fijuljanin, S. Jouette and R. Chollet, J. Microbiol. Methods, 2014, 99, 71–80 CrossRef PubMed.
- R. Doebler, B. Erwin, A. Hickerson, B. Irvine, D. Woyski, A. Nadim and J. Sterling, JALA, 2009, 14, 119–125 CAS.
- P. Vandeventer, K. Weigel, J. Salazar, B. Erwin, B. Irvine, R. Doebler, A. Nadim, G. Cangelosi and A. Niemz, J Microbiol, 2011, 49, 2533–2539 Search PubMed.
- J. Siegrist, R. Gorkin, M. Bastein, G. Stewart, R. Peytavi, H. Kido, M. Bergeron and M. Madou, Lab Chip, 2010, 10, 363–371 RSC.
- K. Hwang, S. Kwon, S. Jung, H. Lim, W. Jung, C. Park, J. Kim, K. Suh and N. Huh, Lab Chip, 2011, 11, 3649–3655 RSC.
- J. Elizalde, M. Antoñana, L. Matthys, F. Laouenan and J. M. Ruano, In Proc. MicroTAS, 2013 Search PubMed.
- S. Vandecasteele, W. Peetermans, R. Mercks and J. Van Eldere, J Microbiol, 2001, 183, 7094–7101 CAS.
- P. Francois, D. Pillet, M. Bento, B. Pepey, P. Vaudaux, D. Lew and J. Schrenzel, J. Clin. Microbiol., 2003, 41, 254–260 CrossRef CAS.
- B. Gillespie and S. P. Oliver, J. Dairy Sci., 2005, 88, 3510–3518 CrossRef CAS.
- M. Hohnadel, L. Felden, D. Fijuljanin, S. Jouette and R. Choued, J. Microbiol. Methods, 2014, 99, 71–80 CrossRef CAS PubMed.
- R. Nandakumar, A. Gounot and B. Mattiasson, J. Biotechnol., 2000, 83, 211–217 CrossRef CAS.
- P. Verhoeven, F. Grattard, A. Carricajo, B. Pozzetto and P. Berthelot, J. Clin. Microbiol., 2010, 48, 4242–4244 CrossRef PubMed.
- M. Smieja, S. Castriciano, S. Carruthers, G. So, S. Chong, K. Luissantra, J. Mahony, A. Petrich, M. Chernesky, M. Savarese and D. Triva, J. Clin. Microbiol., 2010, 18, 3340–3342 CrossRef PubMed.
- G. Kenneth van Horn, C. D. Audette, K. A. Tucker and D. Sebeck, Diagn. Microbiol. Infect. Dis., 2008, 62, 471–473 CrossRef PubMed.
- P. Wilson, M. Labonte, J. Russell, S. Louie, A. Ghobrial and R. Ladner, Nucleic Acids Res., 2011, 39, 17 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |