
Controllable and mass fabrication of highly luminescent N-doped carbon dots for bioimaging applications†
Abstract
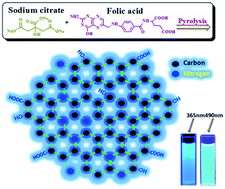
A facile but effective bottom-up method for the mass preparation of N-doped carbon dots (N–C-dots) was developed by the direct heating of a solid mixture of folic acid (FA) and sodium citrate (SC) for several minutes at 300 °C. The nitrogen content of the resulting N–C-dots could be easily and precisely tuned from 0% to 14% by adjusting the SC to FA mass ratio. Uniform N–C-dots with honeycomb-like ordered structures were obtained, with sizes ranging from 1 to 3 nm in diameter. The N-doping content affected not only the emission quantum yield but also the emission wavelength. The N–C-dots emitted strong blue-green fluorescence based on the excitation wavelength and N-doping content. Because of their excellent water solubility, low toxicity, powerful fluorescence, and high resistance to photobleaching, the N–C-dots can enter various cells and serve as ideal candidates for multicolour cell imaging.
 Please wait while we load your content...
Please wait while we load your content...

