
Persistent luminescent and photocatalytic properties of ZnxGa2O3+x (0.8 ≤ x ≤ 1) phosphors
Darong Li,
Yinhai Wang*,
Ke Xu,
Hui Zhao and
Zhengfa Hu
School of Physics and Optoelectronic Engineering, Guangdong University of Technology, Waihuan Xi Road, No.100, Guangzhou 510006, People's Republic of China. E-mail: yhwang@gdut.edu.cn; Fax: +86-20-39322265
First published on 18th February 2015
Abstract
The ZnxGa2O3+x (x = 1, 0.95, 0.9, 0.85, 0.8) phosphors were synthesised successfully via high temperature solid-state reaction. The photoluminescent, persistent luminescent and photocatalytic properties of the phosphors had been studied systematically. The results indicated that their excitation and emission spectra were similar to those of ZnGa2O4 phosphors and all of them have excellent persistent luminescent and photocatalytic properties. The optical properties were changed with the ratio of the Zn2+/Ga3+ and the Zn0.85Ga2O3.85 phosphor showed the best persistent luminescent and photocatalytic properties. The Zn0.85Ga2O3.85 phosphor can be effectively activated by an ultraviolet lamp and ultraviolet excitation can lead to 10 min of persistent green emission. Moreover, the Zn0.85Ga2O3.85 could provide more defect energy levels which can act as photogenerated electron traps and maintain the electron–hole pairs for a longer period, enhancing the persistent luminescent and photocatalytic performance.
1. Introduction
Persistent luminescence is an optical phenomenon, whereby a material is excited with radiation and resulting in luminescent emission remains visible for a long time (seconds to hours) after the excitation has stopped.1–3 These materials are drawing more and more attention in recent years due to the great potential applications in many fields, such as emergency signage, traffic signs and in vivo bio-imaging.Recently, zinc gallate (ZnGa2O4) phosphor has attracted much attention for use in field emission display (FED) and thin film electroluminescence device (TFED), because ZnGa2O4 phosphor shows higher chemical and thermal stability than sulfide phosphors.1,2 ZnGa2O4 crystallizes in the normal spinel structure (Fd3m) with Ga3+ ions occupying octahedral sites and Zn2+ ions occupying tetrahedral sites. Its band gap is about 4.4 eV, and it exhibits a strong blue emission for material not doped with impurities.2 ZnGa2O4 can also act as a host material for multicolor emitting phosphor when doped with transition metals: Cr-doped ZnGa2O4 (ZnGa2O4:Cr3+) for red emission3 and Co-doped ZnGa2O4 (ZnGa2O4:Co3+) for reddish orange emission.4 In addition, ZnGa2O4 has excellent performance in air-pollution control. Because ZnGa2O4 has hybridized orbitals of Zn4s4p, Ga4s4p and the wide band gap (4.4 eV), it can improve the mobility of photogenerated electrons and the absorption efficiency in ultraviolet (UV) lamps.5 Some papers reported that high temperature reaction triggered the vaporization of zinc ions and formed intrinsic defect in the hosts.4,5 Thus, ZnGa2O4 exhibits the persistent luminescent characteristics. However, the influence of zinc deficiency on the persistent luminescent and photocatalytic performance of ZnGa2O4 phosphor is rarely reported.
In this study, we synthesised the ZnGa2O4 phosphor with slightly Zn-deficient composition successfully and investigated their effects on the luminescent, persistent luminescent and photocatalytic properties. The mechanism for enhancement of persistent luminescent and photocatalytic properties was also discussed.
2. Experimental
The ZnGa2O4 phosphors were synthesised via high temperature solid-state reaction method. ZnO powder (99.9%), Ga2O3 powder (99.9%) were milled for about 30 min in an agate mortar. The stoichiometric proportions of raw materials were weighed according to the nominal compositions of ZnGa2O4 (S1), Zn0.95Ga2O3.95 (S2), Zn0.9Ga2O3.9 (S3), Zn0.85Ga2O3.85 (S4), Zn0.8Ga2O3.8 (S5), respectively. Then the mixtures were moved into a corundum crucible and sintered at 1300 °C for 5 hours in an air ambient.The X-ray powder diffraction (XRD) verified the phase structure of all samples by using a diffractometer with Cu Kα irradiation (λ = 1.5406 Å) at 36 KV tube voltage and 20 mA tube current. We set the scanning range from 10° to 70°. The excitation, emission spectra and afterglow decay were measured by a Hitachi F-7000 Florescence Spectrophotometer. Prior to the afterglow decay measurements, the samples were excited by an UV lamp (λex = 254 nm) for 5 min. Then measurement was in a dark place without excitation. The thermoluminescent (TL) curves were recorded by a FJ427A1 thermoluminescent dosimeter. The heating rate was 1 °C s−1 for all samples. Prior to the TL measurement, each sample was first exposed to an UV lamp (λex = 254 nm) for 2 min and then put in dark waiting for 3 min.
The photocatalytic degradation tests were carried out in photoreactor (BL-GHX-V), using 50 mg S1, S2, S3, S4 and S5 powder dispersed in 200 mL Rhodamine B (RhB) aqueous solution (10 mg L−1), respectively. The mixture aqueous solution was put into a beaker with magnetic stirring for 30 min. The light source was a 500 W UV lamp and continuous magnetic stirring should be maintained the suspension of powder in the RhB solution. Then 5 mL of the suspension was gathered for every 10 min and measured via an Ultraviolet-visible Light Spectrometer by measuring its absorbance at a wavelength of 550 nm.
3. Results and discussion
The XRD patterns of five samples are shown in Fig. 1. The diffraction peaks of S1 can be indexed as a pure cubic spinel (space group Fd3m) phase ZnGa2O4, and all diffraction peaks are in good agreement with the standard data of ZnGa2O4. For S4 and S5, extra peaks are appeared in addition to the peaks of ZnGa2O4, which correspond to the phase of β-Ga2O3. It reveals that the obtained products are the mixture of ZnGa2O4, β-Ga2O3 and ZnxGa2O3+x. Compared with the standard data of JCPDS no. 38-1240, there is no apparent shift can be found in all samples. | ||
| Fig. 1 XRD patterns of all samples. | ||
As a kind of self-activated phosphor, ZnGa2O4 emits blue emission. Fig. 2a shows the excitation and emission spectra of ZnxGa2O3+x (x = 1, 0.95, 0.9, 0.85, 0.8) phosphors. The excitation spectrum monitored at 430 nm shows a broad band from 230 nm to 280 nm with the peak position at 254 nm, which can be attributed to the charge transfer of Ga–O.2 Under excitation with 254 nm UV, the emission spectrum of all samples consists of a broad band ranging from 390 nm to 520 nm with a maximum at 430 nm, which is originated from the self-activation center of the octahedral Ga–O group in the ZnGa2O4 lattices.2,5 Moreover, there is a weak emission peak at 500 nm which can be attributed to the 2EA → 4A2 transition of Ga3+ in the distorted octahedral.2,5 The red shift phenomenon in the emission with the decrease of Zn2+ concentration is observed. Because the ionic radii of the Ga3+ is greater than Zn2+, the radii of the ions are as follows: R(Zn2+) = 0.060 nm, R(Ga3+) = 0.062 nm.6 The Zn2+ substituted by the Ga3+ increases the lattice constant of ZnGa2O4. Thus, the peak of emission shifts toward long wavelength. The decay curves of five samples recorded within 0–600 s are presented in Fig. 2b. As can be seen, all samples exhibit similar decay processes which contain a rapid decay at beginning and a slow decay process afterward.7,8 The sequence of the initial intensity for different Zn2+ concentration is x = 0.85 > x = 0.9 > x = 0.8 > x = 0.95 > x = 1.0. In our study, the decay curves can be well fitted by a double-exponential equation and as follows:

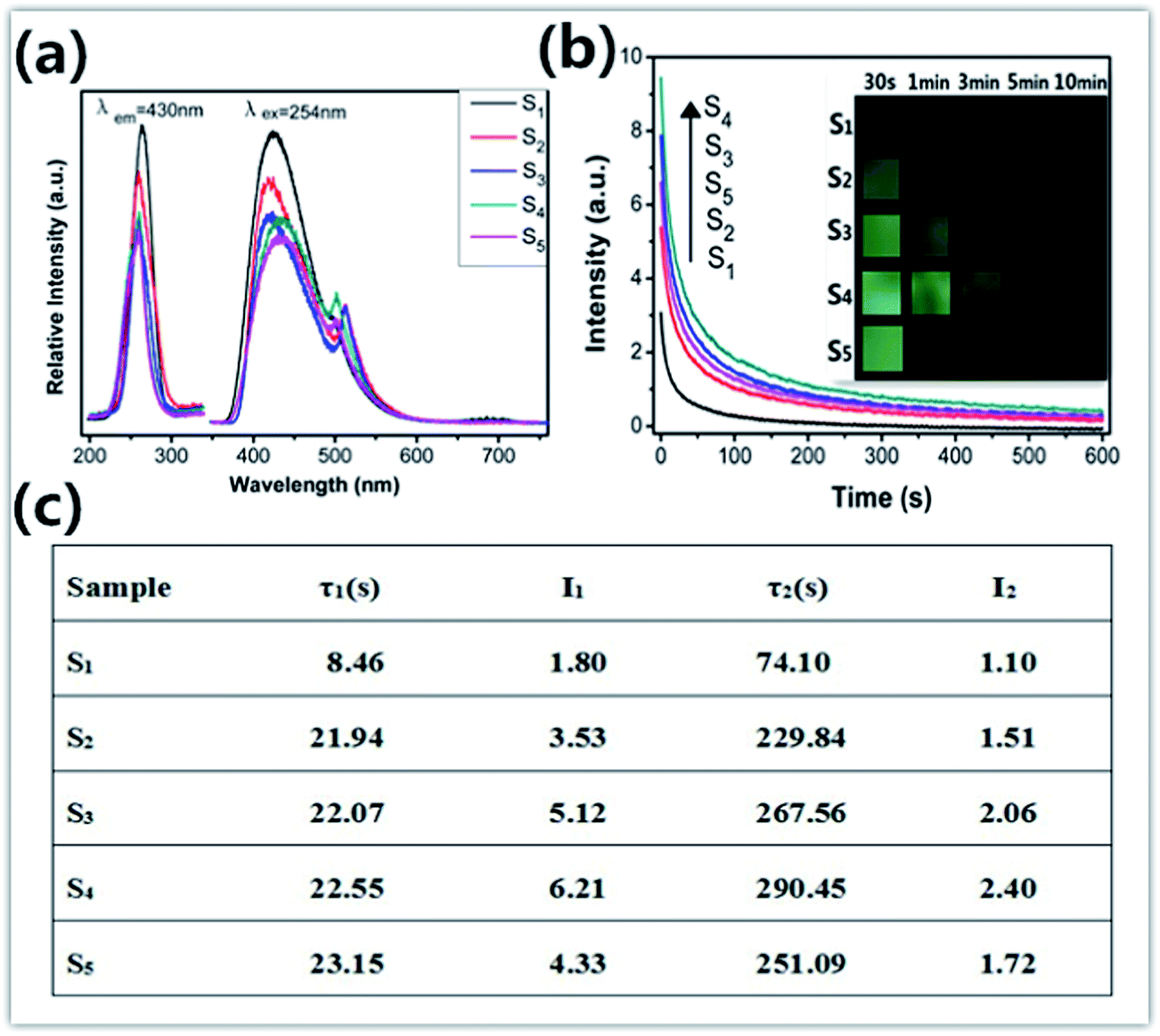 | ||
| Fig. 2 (a) Excitation and emission spectra of all samples; (b) persistent luminescent decay curve after irradiation by 254 nm and images of all samples taken at different persistent luminescence times; (c) the fitting results of decay curves. The phosphors were irradiated for 5 min. | ||
As already known, thermoluminescence (TL) measurement provides an efficient way to reveal the information of traps. In general, the trap concentration is approximately proportional to the TL intensity and the maximum temperature of the TL peak provides information on the depth of the carrier trap,9–11 and the optimal TL peak is usually located at the temperature range of 50–120 °C for the excellent persistent luminescent properties.9,11 As shown in Fig. 3, S1 shows a quite weak TL band centered at 75 °C which confirms the poor persistent luminescence. The TL curves of S2, S3, S4 and S5 have the same peak location and similar shape to that of S1, but their intensity are largely enhanced, and it indicates a higher defect concentration. It is well known that the ZnO is volatilized during ZnGa2O4 synthesis due to the high vapor pressure. One vacancy defect of 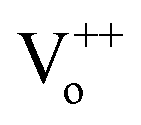 with 2 positive charges and one vacancy defect of
with 2 positive charges and one vacancy defect of  with 2 negative charges would be created in the host. Then the Ga3+ ions would occupy the defect of
with 2 negative charges would be created in the host. Then the Ga3+ ions would occupy the defect of  and from GaZn+, which act as the electron trap. And the defect of
and from GaZn+, which act as the electron trap. And the defect of  would transform into the
would transform into the  and act as the hole trap. Moreover, more Ga3+ ions occupy the
and act as the hole trap. Moreover, more Ga3+ ions occupy the  sites with Zn2+ deficient, which will raise the concentration of the GaZn+ (electron trap). Therefore, the persistent luminescence enhances firstly. However, the distance of Ga–Ga would be shortened as the concentration of GaZn+ continues to raise with Zn2+ deficient, the concentration quenching occurs. Thus, the persistent luminescence begins to decrease.
sites with Zn2+ deficient, which will raise the concentration of the GaZn+ (electron trap). Therefore, the persistent luminescence enhances firstly. However, the distance of Ga–Ga would be shortened as the concentration of GaZn+ continues to raise with Zn2+ deficient, the concentration quenching occurs. Thus, the persistent luminescence begins to decrease.
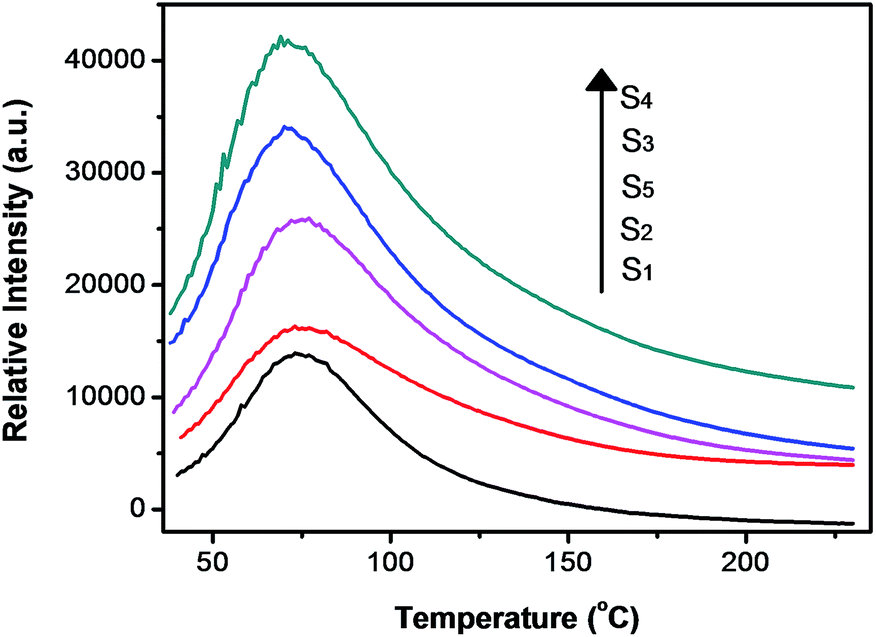 | ||
| Fig. 3 TL curves of all samples. | ||
Fig. 4 shows photocatalytic activity as a function of irradiation time with different Zn2+ ion concentration. It can be seen that with the decrease of Zn2+ concentration, the photocatalytic activities of ZnxGa2O3+x (x = 1, 0.95, 0.9, 0.85, 0.8) first increase and then decrease. The Zn0.85Ga2O3.85 phosphor exhibits the highest photocatalytic activity, and the decomposition rate for the S4/Rhodamine B (RhB) composites reached 98.5% after 90 min. As we known, the high density of surface hydroxyls, proper band positions, broad hybridized orbitals, and large surface area all would improve the high photocatalytic activity of ZnGa2O4.12 And ZnGa2O4 could provide more defect energy levels which can act as photogenerated electrons trap and maintain the electron–hole pairs for a while, enhancing the photocatalytic performance.13,14 In our case, a large numbers of oxygen vacancies have been found in ZnGa2O4 phosphors as evident from TL results. The oxygen vacancies act as an electron traps, which implies a decreased recombination rate of the electrons and holes. The large numbers of oxygen vacancies generate more hydroxyl radicals on the surface, and the strong redox ability of photogenerated electron–hole pairs may also improve the photocatalytic performance. The UV-Vis diffuse reflectance spectra of ZnxGa2O3+x are displayed in Fig. 5a. And the band gap is determined based on the UV-Vis spectra and is displayed in Fig. 5b. It can be seen that with Zn2+ ion concentration decreases, Eg (band gap energy) of ZnxGa2O3+x decreases, increasing higher UV utility efficiency. Thus, photocatalytic activity is improved. However, the reducing of Eg would make EVB (valence bands) and ECB (conduction band) of ZnxGa2O3+x phosphors shift to the Fermi level and decrease the redox ability of photogenerated electron–hole pairs.15,16 Thus, further decrease of Zn2+ concentration may lead to decrease in photocatalytic performance of ZnGa2O4.
 | ||
| Fig. 4 Degradation of RhB in the presence of all samples. The light source was a 500 W UV lamp. | ||
 | ||
| Fig. 5 (a) The UV-Vis diffuse reflectance spectra; (b) optical band gap Eg of all samples. | ||
4. Conclusion
In summary, the ZnxGa2O3+x (x = 1, 0.95, 0.9, 0.85, 0.8) phosphors were synthesised via high temperature solid-state reaction successfully. The influence of Zn2+ ions concentration on PL, decay curves, TL and photocatalytic activity were studied in detail. The emission intensity decreases and the peak of emission shifts toward long wavelength with the decrease of Zn2+ ion concentration. The Zn0.85Ga2O3.85 phosphor can be effectively activated by an UV lamp and the UV excitation can lead to 10 min of persistent green emission. According to the analysis of TL curves, it can be supposed that more Ga3+ ions occupy the sites with Zn2+ deficient, which will raise the concentration of electron trap. However, the distance of Ga–Ga would be shortened as the concentration of GaZn+ continues to raise, the concentration quenching occurs. Studies of the ZnGa2O4 phosphor photocatalytic activity have indicated that with Zn2+ ion concentration decreases, Eg (band gap energy) of ZnxGa2O3+x decreases, increasing higher UV utility efficiency. And photocatalytic activity is improved. But the reducing of Eg would make EVB and ECB of ZnxGa2O3+x phosphors shift to the Fermi level and decrease the redox ability of photogenerated electron–hole pairs. Therefore, photocatalytic performance first increases and then decreases at the same condition.
sites with Zn2+ deficient, which will raise the concentration of electron trap. However, the distance of Ga–Ga would be shortened as the concentration of GaZn+ continues to raise, the concentration quenching occurs. Studies of the ZnGa2O4 phosphor photocatalytic activity have indicated that with Zn2+ ion concentration decreases, Eg (band gap energy) of ZnxGa2O3+x decreases, increasing higher UV utility efficiency. And photocatalytic activity is improved. But the reducing of Eg would make EVB and ECB of ZnxGa2O3+x phosphors shift to the Fermi level and decrease the redox ability of photogenerated electron–hole pairs. Therefore, photocatalytic performance first increases and then decreases at the same condition.
Acknowledgements
This work is supported by the National Nature Science Foundation of China (no. 21271048) and State Key Laboratory of Rare Earth Resource Utilization Open Projects (RERU2013007).References
- M. Allix, S. Chenu, E. Véron, T. Poumeyrol, E. A. Kouadri-Boudjelthia, S. Alahraché, F. Porcher, D. Massiot and F. Fayon, Chem. Mater., 2013, 25, 1600 CrossRef CAS.
- W. N. Kim, H. L. Park and G. C. Kim, Mater. Lett., 2005, 59, 2433 CrossRef CAS PubMed.
- Y. Zhuang, J. Ueda and S. Tanabe, Appl. Phys. Express, 2013, 6, 052602 CrossRef.
- L. P. Sosmana, T. Abrittaa, A. C. Pereirab and H. Vargasb, Chem. Phys. Lett., 1994, 227, 485 CrossRef.
- W. Zhang, J. Zhang, X. Lan, Z. Chen and T. Wang, Catal. Commun., 2010, 11, 1104 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., 1976, 32, 751 CrossRef.
- P. Huang, D. Liu, C. E. Cui, L. Wang and G. Jiang, Appl. Phys. A, 2014, 116, 759 CrossRef CAS.
- Y. Zhuang, J. Ueda and S. Tanabe, J. Mater. Chem. C, 2013, 1, 7849 RSC.
- Y. Jin, Y. Hu, L. Chen, X. Wang, Z. Mu, G. Ju and T. Wang, Mater. Lett., 2014, 126, 75 CrossRef CAS PubMed.
- X. Xu, X. Zhang, T. Wang, J. Qiu and X. Yu, Mater Lett., 2014, 127, 40 CrossRef CAS PubMed.
- W. Xie, Y. Wang, C. Zou, J. Quan and L. Shao, J. Alloys Compd., 2015, 619, 244 CrossRef CAS PubMed.
- H. Li, S. Yin, Y. Wang, T. Sekino, S. W. Lee and T. Sato, J. Mater. Chem. A, 2013, 1, 1123 CAS.
- W. S. Tung, W. A. Daoud and G. Henrion, Thin Solid Films, 2013, 545, 310 CrossRef CAS PubMed.
- X. Wang, M. Blackford, K. Prince and R. A. Caruso, ACS Appl. Mater. Interfaces, 2012, 4, 476 CAS.
- Y. An, L. Yang, J. Hou, Z. Liu and B. Peng, Opt. Mater., 2014, 36, 1390 CrossRef CAS PubMed.
- H. Li, S. Yin and T. Sato, Appl. Catal., B, 2011, 106, 586 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |