DOI:
10.1039/C5RA01008J
(Paper)
RSC Adv., 2015,
5, 34985-34991
Catalytic polymer reactor with “self-sorting” domains for hierarchical catalysis
Received
18th January 2015
, Accepted 30th March 2015
First published on 31st March 2015
Abstract
This study is aimed at the present challenge in self-controlled catalysis, about how catalysts can be furnished with hierarchical catalytic ability. This issue was addressed by constructing a unique catalytic polymer reactor containing “self-sorting” switchable domains that acted as a molecular switch for providing sequenced access to the encapsulated metal nanoparticles. This polymer reactor showed poor catalytic reactivity at relatively low temperatures due to the closed access in the switchable domains, which blocked substrate from the catalytic metal nanoparticles. This polymer reactor showed, however, significant catalytic reactivity for small molecules of substrate at modest temperatures, arising from relaxing of the access in the switchable domains, which allowed small molecules to gain entrance into the encapsulated metal nanoparticles. This polymer reactor further showed significant reactivity for large molecules of substrate at relatively high temperatures, in response to the opening at the switchable domains. In this way, this catalytic polymer reactor demonstrated a hierarchical catalytic ability. This suggested protocol opens up the opportunity to develop smart catalysts for controlled chemical processes.
1. Introduction
Despite the tantalizing prospect in chemical synthesis and in inaccessible places, hierarchical catalysis remains still a significant challenge. At the forefront of this field would be the use of catalytic polymer reactors to achieve self-controlled catalytic ability. From the earliest endeavors, exemplified by poly(N-isopropylacrylamide) (PNIPAm)-encapsulated Ag nanoparticles,1,2 catalytic polymer reactors have been shown to demonstrate switchable catalytic ability in water. This result arises from the temperature-dependent hydrophilic/hydrophobic transition in PNIPAm, which leads to either impeded or unobstructed access into the encapsulated Ag nanoparticles. In this way, catalysis by the polymer reactors demonstrates a switchable process. Impressive progress has been made in this field by adopting a variety of functional polymers as the support of metal nanoparticles. These functional polymers, including smart hydrogels,3 core–shell architectures4 and functional microspheres,5 often have self-regulated properties in controlling the reactants' access, which make feasible the switchable catalysis. Nonetheless, the development of catalytic polymer reactors has not reached its full potential, given the fact that most of the practical catalytic processes involve multi-component reactants and multi-reaction processes,6–8 which desire polymer reactors that have hierarchical catalytic ability and can be made to switch for two or more chemical processes. Unfortunately, it is not realistic to directly acquire such polymer reactors basing on currently available methods and technology. As such, new methods and technology are needed.
For centuries, mankind has been learning and achieving knowledge from nature. A body of knowledge is already available. One of these is the recent recognition of “self-sorting” at bio-polymers,9,10 which appears to share a promising prospect with the stranded field of catalytic polymer reactors. Consisting of diverse amino acids that engage in a broad range of interactions, the bio-polymers, such as protein, can form ordered self-assembled architectures that autonomously adapt to the change of environments. The main reason behind this can be related to the self-sorting of these complicated interactions, where the relatively stronger interactions may greater dictate the polymeric conformations while the relatively weaker interactions provide a synergy. In this manner, the self-sorting effect endows the polymer with environment's adaptability. A rapid change in the environment would induce a stepwise dissociation/association at these interactions, resulting in sequenced switching of the polymeric conformations. In this way, the self-sorting effect allows for the occurrence of controllable behaviors. Although these reported works are not directly related to catalytic applications, the method developed in the self-sorting field provides a new insight into the stranded field of polymer reactors, which makes feasible sequenced switchable domains.
Inspired by this principle, herein we aimed at the present challenge in self-controlled catalysis by constructing a unique catalytic polymer reactor furnished with “self-sorting” domains. This polymer reactor (named “NiPR-HCS”) was fabricated with nickel nanoparticles and a polymer composite containing “self-sorting” domains made of two different interactions, that is, weak hydrogen bonds (between PAAm and PTFMA) and relatively stronger polymer complexes (between PVIm and PTFMA) (PAAm: poly(acrylamide); PTFMA: poly(2-trifluoromethylacrylic acid); PVIm: poly(1-vinylimidazole)). The self-sorting switchable domains act as a molecular switch for providing sequenced access to the encapsulated metal nanoparticles. As proposed in Scheme 1, the closed access in the switchable domains at relatively low temperatures intends to block substrate from the encapsulated metal nanoparticles, causing poor catalytic reactivity (cf. Route A). The access for small molecules of substrate is, however, allowed with increasing temperature, arising from disrupting of these hydrogen-bonding interactions (cf. Route B). The access may be opened for a bigger molecule at relatively high temperatures, resulting from the dissociation of the PTFMA–PVIm complexes (cf. Route C). In this way, this catalytic polymer reactor shows the hierarchical catalytic ability. To that end, 4-nitrophenol (4-NP) and Rhodamine B (RB) were selected as the tentative substrates, given their difference in molecular sizes (RB much larger than 4-NP) and the fact that their reductions with borohydrides are the common model reactions in catalytic test.11,12 The objective of this study is to demonstrate that catalytic polymer reactors that have hierarchical catalytic ability can be prepared by using this novel protocol, which opens up the opportunity to develop smart catalysts for controlled chemical processes.
 |
| | Scheme 1 Proposed mechanism for the NiPR-HCS reactor. | |
2. Experimental section
2.1. Preparation of polymer reactors
Unless otherwise noted, the chemicals used were of analytic grade and used as received from Sigma-Aldrich. This novel polymer reactor, as outlined in Scheme 1, was prepared basing on the suggested design for interpolymer complexes,13,14 where the functional monomers 2-trifluoromethylacrylic acid (TFMA), 1-vinylimidazole (VIm) and acrylamide (AAm) were used in a molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 to ensure the entire complexation between carboxyl groups and both imidazole and amino moieties. In detail, TFMA (0.40 g; 2.8 mmol), VIm (0.13 g; 1.4 mmol) and AAm (0.10 g; 1.4 mmol) are dissolved in dimethyl sulfoxide (10 mL). After being dispersed and deoxygenated with sonication and nitrogen, the initiator ammonium persulfate (0.13 g; 0.57 mmol) and crosslinker N,N′-methylene diacrylamide (0.05 g; 0.34 mmol), along with nickel nitrate hexahydrate (0.21 g; 0.7 mmol), were added. The mixture system was subsequently irradiated with ultraviolet light (365 nm) overnight. The encapsulated ionic nickel was reduced by an excess of sodium borohydride (tenfold, with regard to ionic nickel). The resulted block polymer was crushed and grinded into a size of ca. 60 mesh, washed with ethanol and water, and then dried under flowing nitrogen. In this way, this polymer reactor (i.e., NiPR-HCS) was prepared.
:1:1 to ensure the entire complexation between carboxyl groups and both imidazole and amino moieties. In detail, TFMA (0.40 g; 2.8 mmol), VIm (0.13 g; 1.4 mmol) and AAm (0.10 g; 1.4 mmol) are dissolved in dimethyl sulfoxide (10 mL). After being dispersed and deoxygenated with sonication and nitrogen, the initiator ammonium persulfate (0.13 g; 0.57 mmol) and crosslinker N,N′-methylene diacrylamide (0.05 g; 0.34 mmol), along with nickel nitrate hexahydrate (0.21 g; 0.7 mmol), were added. The mixture system was subsequently irradiated with ultraviolet light (365 nm) overnight. The encapsulated ionic nickel was reduced by an excess of sodium borohydride (tenfold, with regard to ionic nickel). The resulted block polymer was crushed and grinded into a size of ca. 60 mesh, washed with ethanol and water, and then dried under flowing nitrogen. In this way, this polymer reactor (i.e., NiPR-HCS) was prepared.
For a contrastive study, four controls, named “NiPR-N”, “NiPR-HS”, “NiPR-CS” and “PR-HCS”, respectively, were also prepared under comparable conditions. NiPR-N was the non-responsive nickel reactor where the polymeric carrier was made entirely of PVIm (herein, the ‘N’ suffix means the ‘non-responsive’ properties in contrast to the ‘S’, switchable characteristics in other polymer reactors). PR-HCS was the polymeric carrier of NiPR-HCS and prepared without using nickel. NiPR-HS was the switchable nickel reactor made of PAAm and PTFMA (i.e., containing hydrogen bonds, only) and prepared in the absence of VIm. NiPR-CS was the switchable nickel reactor comprising PVIm–PTFMA complexes and prepared without using AAm (herein, the ‘HS’ and ‘CS’ suffixes in NiPR-HS and NiPR-CS denote the hydrogen-bonding responsiveness and the polymeric complexes, respectively, in contrast to the combined switching in NiPR-HCS). For a convenient discussion, all of the prepared polymer reactors and carrier were mentioned henceforth as the conceptual catalytic reactors.
2.2. TEM and FT-IR analysis
The TEM images of the above-prepared reactors are obtained using a transmission electron microscope (TEM) (JEM-2100, Japan). The infrared spectra were recorded using a FT-IR apparatus (Nicolet MX-1E, USA).
2.3. Self-switchable interactions
The self-switchable interactions between PAAm and PTFMA and between PVIm and PTFMA were studied as a function of temperature, by using dynamic light scattering (DLS) (Bettersize 2000, China). For equilibrium, all the samples concerned were kept at the specified temperatures for at least 10 min before acquiring hydrodynamic radii (Rh). By a comparison between the switchable reactors and the non-responsive NiPR-N, the contribution of the switchable interactions was therefore reflected by the change of swelling ratio (Rc):15
Herein, Rd is the particle size of dried particles, ‘S’ means the switchable reactors and ‘N’ represents the non-responsive reactor.
2.4. Catalysis test
The catalytic properties of the prepared polymer reactors were evaluated in batch format at room temperature.11,12 The substrate (4-NP or RB) was added into NaBH4 aqueous solution with the initial concentration 0.01 μmoL mL−1 (total volume: 3 mL) (NaBH4, 20 folds with regard to the substrate). The solid content of the polymer reactors used in every test was 0.06 mg mL−1. The reduction of the substrate was monitored spectrophotometrically. The catalytic reactivity of these polymer reactors were obtained from the average of three runs.
2.5. Electrochemical tests
Electrochemical tests were further performed to interrogate the catalytic mechanism between the prepared polymer reactors and substrate.16 Using an electrochemical workstation furnished with a three-electrode configuration (Au-plate working electrode, Pt-wire counter electrode and Ag/AgCl ref. electrode) (CHI760E, China), polymer reactors (10 mg) that pre-absorbed ca. 10 nmol substrate (i.e., 4-NP and RB) were placed into a cuvette encircled by a diffusion-eliminating sonication apparatus (supporting electrolyte: 0.01 mmol mL−1 KCl; 10 mL). The substrate desorbed transiently was cyclic-voltammetrically scanned with the workstation until a stable desorption/reduction profile was achieved (scanning range, 0–−0.8 V; rate, 1 mV s−1).
3. Results and discussion
3.1. FT-IR and SEM analysis
The polymer reactor NiPR-HCS, as aforementioned, was constructed from nickel nanoparticles and a unique polymer composite of PTFMA, PVIm and PAAm. FT-IR was first used to characterize the composition, as shown in Fig. 1. Three major bands (i.e., 3000–3750, ∼1750 and 1000–1500 cm−1) appeared in the FT-IR spectrum of NiPR-HCS, a result corresponding to the vibrating zones of O–H (N–H), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N (C–C), respectively.17 Given the constituent PTFMA, PVIm and PAAm, plus the crosslinker, which separately contains these vibrational groups, it is therefore difficult to identify one specified component from NiPR-HCS. As such, we also included the FT-IR spectra of four controls described above (i.e., NiPR-N, NiPR-HS, NiPR-CS and PR-HCS, made of PVIm, PTFMA–PAAm, PTFMA–PVIm, and PTFMA–PVIm–PAAm, respectively) in Fig. 1. NiPR-HCS exhibited almost the same spectrum as PR-HCS and included the major absorption bands of these controls. The identical spectra between PR-HCS and NiPR-HCS can be ascribed to the comparable composition between PR-HCS and the polymeric carrier of NiPR-HCS. The inclusion of the major bands of these controls in NiPR-HCS suggests the multi-component copolymer compounded by PTFMA, PVIm, and PAAm. It is therefore clear that the prepared NiPR-HCS was a polymer composite of PTFMA, PVIm, and PAAm, as expected. Fig. 2 presents the TEM images exhibiting the morphology of metal nanoparticles encapsulated in these polymer reactors. Nickel nanoparticles with a size of ca. 10 nm were encapsulated in the polymeric building blocks. Hence, this novel polymer reactor NiPR-HCS was prepared in the desired form.
O and C–N (C–C), respectively.17 Given the constituent PTFMA, PVIm and PAAm, plus the crosslinker, which separately contains these vibrational groups, it is therefore difficult to identify one specified component from NiPR-HCS. As such, we also included the FT-IR spectra of four controls described above (i.e., NiPR-N, NiPR-HS, NiPR-CS and PR-HCS, made of PVIm, PTFMA–PAAm, PTFMA–PVIm, and PTFMA–PVIm–PAAm, respectively) in Fig. 1. NiPR-HCS exhibited almost the same spectrum as PR-HCS and included the major absorption bands of these controls. The identical spectra between PR-HCS and NiPR-HCS can be ascribed to the comparable composition between PR-HCS and the polymeric carrier of NiPR-HCS. The inclusion of the major bands of these controls in NiPR-HCS suggests the multi-component copolymer compounded by PTFMA, PVIm, and PAAm. It is therefore clear that the prepared NiPR-HCS was a polymer composite of PTFMA, PVIm, and PAAm, as expected. Fig. 2 presents the TEM images exhibiting the morphology of metal nanoparticles encapsulated in these polymer reactors. Nickel nanoparticles with a size of ca. 10 nm were encapsulated in the polymeric building blocks. Hence, this novel polymer reactor NiPR-HCS was prepared in the desired form.
 |
| | Fig. 1 FT-IR spectra of the prepared polymer reactors (for reactors naming, ‘Ni’ means nickel nanoparticles, ‘PR’ suggests polymer reactors, ‘S’ stands for switchable properties, ‘H’ represents hydrogen bonds and ‘C’ indicates polymeric complexes). | |
 |
| | Fig. 2 TEM images of the metal nanoparticles contained in the prepared polymer reactors ((a): NiPR-HCS; (b): NiPR-N; (c): NiPR-HS; (d): NiPR-CS; (e): PR-HCS). | |
3.2. Evaluation of the self-switchable interactions
Fig. 3 presents the DLS curves of the prepared polymer reactors, with the purpose of confirming the self-switchable interactions. Compared with NiPR-N, the non-responsive reactor, the swelling ratio Rc in the switchable reactors showed significant dependence upon the temperature. The maximal values of Rc in NiPR-HS and NiPR-CS appeared at ca. 30 °C and 45 °C, respectively. Below the temperatures, NiPR-HS and NiPR-CS showed a low Rc associating with the complementary interactions between PAAm and PTFMA (hydrogen bonds) and between PVIm and PTFMA (polymeric complexes), which inhibited swelling of the polymers. Above the temperatures, NiPR-HS and NiPR-CS showed a dramatically increased Rc in response to the dissociation of both interactions. In contrast, the swelling profiles in NiPR-HCS and PR-HCS showed a combined feature between NiPR-HS and NiPR-CS, where the Rc values underwent an initial increase at ca. 30 °C and then another increase at 45 °C. It is therefore suggested that the expected sequenced responsiveness was already incorporated into this novel NiPR-HCS. In conjunction with the development of this polymer reactor (cf. Scheme 1), the sequenced responsiveness potentially makes feasible hierarchical catalytic ability.
 |
| | Fig. 3 DLS curves of the prepared polymer reactors. | |
3.3. Hierarchical catalytic ability
The catalytic properties of the polymer reactors are presented in Fig. 4. Three representative temperatures, i.e., 20, 35 and 50 °C, either higher or lower than the transition temperatures of NiPR-HCS (i.e., 30 and 45 °C) (cf. Fig. 3), were selected in order to scrutinize the switchable catalytic ability. The selection of such temperatures was to ensure that the switchable zones associating with Routes A, B and C can be fully covered (cf. Scheme 1). As shown in Fig. 4, the catalytic reactivity of NiPR-N increased with increasing temperature, regardless of the specified catalytic cases (4-NP or RB). In contrast, PR-HCS did not exhibit essential catalytic ability due to the lack of catalytic nickel nanoparticles. The catalytic reactivity of NiPR-HCS underwent an initial increase for 4-NP (Fig. 4b) and then another increase for RB (Fig. 4c). The catalytic reactivity of NiPR-CS was, however, subject to a major increase at 50 °C. Specifically, at 20 °C, NiPR-HCS, NiPR-HS and NiPR-CS showed poor catalytic reactivity for both 4-NP and RB (lower than NiPR-N) (Fig. 4a). At 35 °C, NiPR-HCS and NiPR-HS showed significant reactivity for 4-NP (higher than NiPR-N) (Fig. 4b). NiPR-HCS further showed significant reactivity for RB at 50 °C (Fig. 4c). The main catalytic reactivity of NiPR-CS came, however, only at 50 °C. The catalytic behavior at NiPR-HCS clearly showed a combined feature between NiPR-HS and NiPR-CS. As desired, NiPR-HCS demonstrated the hierarchical catalytic ability.
 |
| | Fig. 4 Catalytic reactivity of the prepared polymer reactors ((a): 20 °C; (b): 35 °C; (c): 50 °C) (for reference, the catalysis of NiPR-N was denoted with a circlet). | |
3.4. Dynamical binding behavior and switchable access
For electrochemical studies on polymer reactors and their catalytic mechanisms,18 the underlying issue lies with the interaction between the reactors and substrate. It is known that the potential to reduce or oxidize a binding molecule depends upon the binding constant. Stronger binding will need more energy to overcome the binding. As shown in Scheme 2, the substrate (B) in the system would normally involve desorption, diffusion to the surface of the electrodes, and terminal reaction. The overall reaction rate is therefore determined by the slowest step, i.e., the rate-determining step. In the event that the diffusion is eliminated with sonication, the overall reaction rate would directly associate with the terminal reaction. By using thermodynamic theories, the chemical potential of the substrate in bulk solution can be:| |
 | (1) |
where μφ and Cφ are the standard chemical potential of the substrate and the corresponding standard concentration, and C1 is the practical concentration. R is the gas constant (8.314 J mol−1 K−1) and T is the reaction temperature. The correlation of eqn (1) with the absorption/desorption equilibrium of the substrate will give eqn (2):| |
 | (2) |
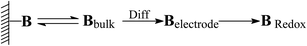 |
| | Scheme 2 Schematic presentation of an electrochemical process with binding molecule B. | |
Herein, the superscript and subscript ‘n’ represent the polymer reactors, implicating the interaction between the reactors and the substrate. K is the equilibrium constant of absorbing the substrate onto the polymer reactors, suggesting the affinity of the polymer reactors towards the substrate. The use of thermodynamic theories on the surface of the working electrode will achieve eqn (3):
| |
 | (3) |
Herein, E is the redox potential of the substrate, N is the number of the electrons transferred in the redox process (in moles), and F is the Faraday constant (96485 C mol−1). The deduction of eqn (2) with eqn (3) leads to eqn (4):
| |
 | (4) |
After eliminating the concentration gradient of the substrate in bulk solution with sonication, eqn (4) can be re-written into eqn (5):
| |
 | (5) |
The consecutive scanning with cyclic voltammetry up to a stable desorption/redox profile would give eqn (6):
| | |
lnK = aE + b
| (6) |
where ‘
a’ and ‘
b’ are constants. The interaction of the substrate with different polymer reactors leads to the expression:
| | |
ΔlnK = aΔE
| (7) |
The binding constant shows direct dependence upon the redox potential. A larger binding constant will result in a higher redox potential. As such, the electrochemical studies were performed in accordance with this paradigm, as shown in Fig. 5. Given the sequenced responsiveness at NiPR-HCS, we further selected 20, 35 and 50 °C for a contrastive study. 4-NP that was attached to NiPR-HCS at 20 °C exhibited a desorption/reduction peak at −501 mV (Fig. 5a). In contrast, this peek at 35 °C shifted to a smaller position (−478 mV) (Fig. 5b). There was not essential shift appearing in this peak at 50 °C, in contrast to that at 35 °C (−475 mV vs. −478 mV; Fig. 5c). The result was reversed for RB, where RB that was attached to NiPR-HCS at 20 °C and 35 °C exhibited comparable desorption/reduction potentials (−520 mV vs. −518 mV; Fig. 5d and e). This peek at 50 °C shifted to a smaller position (i.e., −484 mV) (Fig. 5f). NiPR-HCS showed a stronger interaction with 4-NP at relatively low temperatures yet a stronger interaction with RB at relatively higher temperatures. This result suggests the presence of sequenced access in NiPR-HCS, which was available first to 4-NP and then to RB.
 |
| | Fig. 5 Reduction profiles with substrate desorbing from NiPR-HCS ((a): 4-NP at 20 °C; (b): 4-NP at 35 °C; (c): 4-NP at 50 °C; (d): RB at 20 °C; (e): RB at 35 °C; (f): RB at 50 °C). | |
For further addressing the sequenced access, Table 1 provides the desorption/reduction potentials achieved from all the prepared polymer reactors. Despite the encapsulated metal nanoparticles, NiPR-HCS showed comparable potential with PR-HCS. The potentials at NiPR-HCS and NiPR-HS underwent an initial shift for 4-NP and then another shift for RB. The potential at NiPR-CS was, however, subject to the major shift at 50 °C. In conjunction with the catalytic studies (Fig. 4), this result further suggests that the hierarchical catalytic ability in NiPR-HCS lies with the switchable polymer carrier, which induced sequenced access in NiPR-HCS and thereby the hierarchical catalytic ability.
Table 1 Reduction potentials with substrate desorbing from all the prepared polymer reactors (mV)
| Polymer reactor |
20 °C |
35 °C |
50 °C |
| 4-NP |
RB |
4-NP |
RB |
4-NP |
RB |
| NiPR-HCS |
−501 |
−520 |
−478 |
−518 |
−475 |
−484 |
| PR-HCS |
−499 |
−519 |
−476 |
−517 |
−475 |
−483 |
| NiPR-HS |
−492 |
−501 |
−470 |
−500 |
−470 |
−481 |
| NiPR-CS |
−512 |
−522 |
−510 |
−520 |
−477 |
−485 |
| NiPR-N |
−481 |
−490 |
−480 |
−489 |
−479 |
−487 |
4. Conclusions
This study aimed at the present challenge in self-controlled catalysis by constructing a unique catalytic polymer reactor containing “self-sorting” domains that acted as a molecular switch for providing sequenced access to the encapsulated metal nanoparticles. At relatively low temperatures, this polymer reactor showed poor catalytic reactivity due to the closed access in the switchable domains, which blocked substrate from the catalytic metal nanoparticles. At modest temperatures, this polymer reactor showed, however, significant catalytic reactivity for small molecules of substrate at modest temperatures, arising from relaxing of the access in the switchable domains, which allowed small molecules to gain entrance into the encapsulated metal nanoparticles. At higher temperatures, this polymer reactor further showed significant reactivity for large molecules of substrate, in response to the opening at the switchable domains. It is therefore confirmed that catalytic polymer reactors having hierarchical catalytic ability can be prepared using this novel “self-sorting”-based protocol. Future development in this field will significantly increase the applications, and will lead to the appearance of novel functional catalysts and catalytic materials.
Acknowledgements
The authors want to express their gratitude to the National Science Foundation of China (no. 51473070, 21403091 and 51402128). Thanks also should be expressed to the Jiangsu Province and Jiangsu University for support under both the distinguished professorship program (Sujiaoshi [2012]34, no. 12JDG001) and the innovation/entrepreneurship program (Suzutong [2012]19). The authors also appreciate the project support from the Jiangsu Provincial Science and Technology Agency (BK20130486).
References
- Y. Lu, Y. Mei, M. Drechsler and M. Ballauff, Angew. Chem., Int. Ed., 2006, 45, 813–816 CrossRef PubMed.
- C.-W. Chen, M.-Q. Chen, T. Serizawa and M. Akashi, Adv. Mater., 1998, 10, 1122–1126 CrossRef.
- N. Sahiner, Prog. Polym. Sci., 2013, 38, 1329–1356 CrossRef PubMed.
- Q. Zhang, I. Lee, J. B. Joo, F. Zaera and Y. Yin, Acc. Chem. Res., 2013, 46, 1816–1824 CrossRef PubMed.
- B. Liu, W. Zhang, F. Yang, H. Feng and X. Yang, J. Phys. Chem. C, 2011, 115, 15875–15884 CAS.
- A. Dhakshinamoorthy and H. Garcia, ChemSusChem, 2014, 7, 2392–2410 CrossRef PubMed.
- Y. Lin, Z. Li, Z. Chen, J. Ren and X. Qu, Biomaterials, 2013, 34, 2600–2610 CrossRef PubMed.
- J. Zhou and B. List, J. Am. Chem. Soc., 2007, 129, 7498–7499 CrossRef PubMed.
- Z. Huang, L. Yang, Y. Liu, Z. Wang, O. A. Scherman and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 5351–5355 CrossRef PubMed.
- S. Dong, X. Yan, B. Zheng, J. Chen, X. Ding, Y. Yu, D. Xu, M. Zhang and F. Huang, Chem.–Eur. J., 2012, 18, 4195–4199 CrossRef PubMed.
- S. Li and S. Gong, Adv. Funct. Mater., 2009, 19, 2601–2606 CrossRef PubMed.
- H. P. Singh, N. Gupta, S. K. Sharma and R. K. Sharma, Colloids Surf., A, 2013, 416, 43–50 CrossRef PubMed.
- B. Peng, X. Yuan, M. Zhu and S. Li, Polym. Chem., 2014, 5, 562–566 RSC.
- S. Li, Y. Ge, A. Tiwari and S. Cao, Small, 2010, 6(21), 2453–2459 CrossRef PubMed.
- S. Li, Y. Ge, S. A. Piletsky and A. P. F. Turner, Adv. Funct. Mater., 2011, 21, 3344–3349 CrossRef PubMed.
- Y. Zhou, M. Zhu and S. Li, J. Mater. Chem. A, 2014, 2, 6834–6839 Search PubMed.
- W. Wang, X. Tian, Y. Feng, B. Cao, W. Yang and L. Zhang, Ind. Eng. Chem. Res., 2010, 49, 1684–1690 CrossRef.
- S. Li, Y. Luo, M. Whitcombe and S. A. Piletsky, J. Mater. Chem. A, 2013, 1, 15102–15109 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 












