
The ground-state structure and physical properties of ReB3 and IrB3 predicted from first principles
Abstract
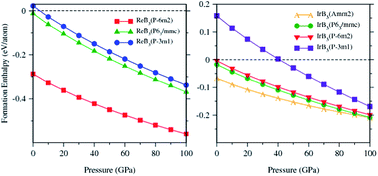
ReB3 has been synthesized and was reported to have symmetry of P63/mmc [Acta Chem. Scand. 1960, 14, 733]. However, we find that this structure is not stable due to its positive formation energy. In 2009, IrB1.35 and IrB1.1 were synthesized and were considered to be superhard [Chem. Mater. 2007, 21, 1407; ACS Appl. Mater. Interfaces 2010, 2, 581]. Inspired by these results, we explored the possible crystal structures of ReB3 and IrB3 by using the developed particle swarm optimization algorithm. We predict that P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) m2-ReB3 and Amm2-IrB3 are the ground-state phases of ReB3 and IrB3, respectively. The stability, elastic properties, and electronic structures of the predicted structures were studied by first-principles calculations. The negative calculated formation enthalpies for Pm2-ReB3 and P63/mmc-ReB3 indicate that they are stable and can be synthesized under ambient pressure. Their dynamical stability is confirmed by calculated phonon dispersion curves. The predicted P63/mmc-ReB3 has the highest hardness among these predicted structures. The calculated density of state shows that these predicted structures are metallic. The chemical bonding features of the predicted ReB3 and IrB3 were investigated by analyzing their electronic localization function.
m2-ReB3 and Amm2-IrB3 are the ground-state phases of ReB3 and IrB3, respectively. The stability, elastic properties, and electronic structures of the predicted structures were studied by first-principles calculations. The negative calculated formation enthalpies for Pm2-ReB3 and P63/mmc-ReB3 indicate that they are stable and can be synthesized under ambient pressure. Their dynamical stability is confirmed by calculated phonon dispersion curves. The predicted P63/mmc-ReB3 has the highest hardness among these predicted structures. The calculated density of state shows that these predicted structures are metallic. The chemical bonding features of the predicted ReB3 and IrB3 were investigated by analyzing their electronic localization function.
 Please wait while we load your content...
Please wait while we load your content...

