DOI:
10.1039/C5RA02063H
(Paper)
RSC Adv., 2015,
5, 25911-25918
Cobalt(II) and copper(II) supramolecular networks with a 1-iminoisoindoline asymmetric pincer†
Received
2nd February 2015
, Accepted 3rd March 2015
First published on 3rd March 2015
Abstract
The first synthesis and characterization of cobalt(II) and copper(II) complexes with an in situ prepared novel asymmetric trinitrogen isoindoline-based pincer type ligand, N-(2-picolyl)isoindoline-1-(2-picolyl)imine (pap), are reported. Single-crystal X-ray structures of the cis-[M(pap)Cl2] (M = Cu, Co) complexes reveal the presence of rare seven-membered chelate rings as well as various coordination geometries and packing modes. The water-soluble complexes were found to react with dicarboxylate ions, terephthalate (ta) and succinate (suc), forming three new compounds {[M(H2O)(pap)]2(μ-ta)}·ta·xH2O (M = Cu and Co) and {[Co(pap)(suc)]·xH2O}n. Their single-crystal X-ray structures are reported and discussed. The rigid ta ions facilitate the formation of binuclear hydrated supramolecular frameworks, whereas the flexible suc linker binds [Co(pap)]2+ moieties into 1D zigzag chains. The chains are non-covalently organized into a supramolecular network. The bulky pincer ligand, responsible for the luminescent properties, was found to be inert in these reactions and to facilitate a unique supramolecular architecture through π–π stacking interactions. The compounds presented in this work may have wider applicability, as molecular building blocks, in the construction of either discrete polynuclear clusters or extended networks with desirable properties.
Introduction
Compounds based on metals and pincer ligands have been known1 since 1976 and after being labelled as academic curiosities they are now considered an important branch of coordination chemistry, having vast applicability in homogenous catalysis and organometallic chemistry.2 Different types of pincers3 were synthesized and characterized including E,C,E type pincers with various E donors, including P, N and S atoms and C-ipso carbanions.4 The majority of these compounds have been obtained in direct one-pot reactions in solution; and a few other approaches, such as post-synthetic modification and mechanochemistry, have been recently reported.5–8 Usually, rigid and bulky analogues of E,C,E pincer ligands coordinate meridionally9 and substitution of the C-ipso atom is associated with gaining non-innocent character by the ligand.10 Another advantage of the pincer ligands is nowadays their asymmetric structure, hemilability and chirality which are the features required for asymmetric catalysis.11,12 Among N,N,N type pincer ligands, those with symmetric 1,3-bis-(arylimino)isoindoline are investigated in catalytical processes due to simplicity of synthesis, ease of deprotonation, robustness, preferential meridional coordination to metal center and stabilization of its certain valence. However, it is worth to note that only a few published papers describe asymmetric 1,2-bis-(R-imino)isoindoline based pincer complexes.13,14 Interestingly, asymmetric 1-iminoisoindoline (1-ISND) pincer chelates have not been yet described and structurally characterized, despite various possible synthetic routes for obtaining 1-ISND derivatives. One of them uses o-phthalaldehyde as a precursor in the synthesis of 1-ISND. It is also commonly employed in analytical chemistry as a fluorescent probe in determination of amino acids content in environmental samples.15 However, the mechanism of formation of fluorescent species as well as their molecular structure have not been yet established. Variety of possibly fluorophoric intermediates in aqueous systems formed during either stepwise formation of cyclic and acyclic hydrates of o-phthalaldehyde or stepwise Schiff base condensation yielding isoindolinone and 1-ISND makes this interpretation difficult.15
The 1-ISND coordination chemistry has huge potential as these bulky building blocks may possibly play an important role in crystal engineering and rational design of porous frameworks. The only examples of 1-ISND based materials are those in which the ligand acts as an angular linker bridging metal centers16–18 and organometallic palladacycles.19–22 Lindoy et al. constructed porous coordination networks based on 1-ISND angular linker and cadmium or iron cations.16 The palladacycles based on 1-ISND were studied by Foley et al. and were found to be active catalysts in C–C coupling reactions.19,21
Up until now, the literature sources give no examples either of chelating tridentate 1-ISND complexes or characterization of 1-ISND photophysical processes in the solid state.
In this work we describe synthesis and characterization of novel copper(II) (1) and cobalt(II) (2) chelate complexes with asymmetric 1-ISND pincer type ligand, N-(2-picolyl)isoindoline-1-(2-picolyl)imine (pap, Scheme 1) and explore their potential as building blocks in a rational construction of supramolecular networks with rigid as well as flexible dicarboxylate ions. Three new materials, incorporating bulky pap co-ligand, of various nuclearity, were obtained in reactions of 1 and 2 with terephthalate {binuclear compounds 3 (Cu2+) and 4 (Co2+)} and succinate dicarboxylates {polynuclear compound 5 (Co2+)}. We present detailed description of all structures in order to identify and compare supramolecular interactions that lead to versatile networks. We also present the results of spectroscopic investigation, including fluorescent properties, of the pap containing materials in order to identify fluorescent processes inside 1-ISND moiety. In addition, we discuss the influence of linker flexibility on nuclearity of the material.
 |
| | Scheme 1 The structural formula of the pap ligand. | |
Experimental section
Materials and methods
Reagents were purchased from commercial suppliers (Sigma-Aldrich) at high purity (>97%) and utilized in experiments as received. Solvents were purchased from Sigma-Aldrich, except EtOH (92%, Polmos). The absorbance UV-VIS spectra were recorded on a Shimadzu UV-VIS-NIR UV-3600 spectrophotometer. Electronic diffuse reflectance spectra were measured in BaSO4 pellets with BaSO4 as a reference using UV-3600 UV-VIS-NIR spectrophotometer equipped with ISR-260 attachment. The IR spectra were recorded on a Thermo Scientific Nicolet iS5 FT-IR spectrometer equipped with an iD5 diamond ATR attachment. The photoluminescence spectra in solid state were recorded on a Perkin Elmer LS55 spectrofluorimeter and the data were corrected for Rayleigh scattering effects. The magnetic susceptibility measurements were performed on a Sherwood scientific magnetic susceptibility balance at 293 K and no diamagnetic corrections were made. Microanalyses on carbon, hydrogen and nitrogen were performed using an Elementar Vario MICRO Cube elemental analyzer. Powder X-ray diffraction (PXRD) patterns were recorded at room temperature (295 K) on a Rigaku Miniflex 600 diffractometer with Cu-Kα radiation (λ = 1.5418 Å) in a 2θ range from 3° to 60° with a 0.01° step at a scan speed of 2° min−1. TGA analysis was carried out on Mettler Toledo TGA/SDTA 851 with a heating rate of 10 deg min−1 under Ar flow (80 mL min−1). Diffraction data for single crystals of 1 and 2 were collected at 293 K on Oxford Diffraction SuperNova four-circle diffractometer, using Mo-Kα and Cu-Kα radiation sources and graphite monochromator. Diffraction data for single crystals of 3, 4 and 5 were collected at 100 K (3, 4) or 120 K (5) using Bruker–Nonius Kappa CCD four-circle diffractometer, equipped with a Mo-Kα radiation source, graphite monochromator and CryoStream system for measurements at low temperature. Cell refinement and data reduction was performed using firmware.23,24 Positions of all non-hydrogen atoms were determined by direct methods using SIR-97.25 All non-hydrogen atoms were refined anisotropically using weighted full-matrix least-squares on F2. Refinement and further calculations were carried out using SHELXL-97.26 All hydrogen atoms joined to carbon atoms were positioned with an idealized geometry and refined using a riding model with Uiso(H) fixed at 1.5Ueq of C for methyl and at 1.2Ueq of C for other groups. The positions of water molecule hydrogen atoms were found from the difference Fourier map and refined with restrained bond lengths and angles. Uiso(H) were constrained to 1.5Ueq of their home O atom. All used programs are components of the WINGX.27 Graphics and geometrical calculations were carried out by using MERCURY28 and Crystal Explorer.29
Synthesis of 1–5
Synthetic procedures described in detail below were carried out under aerobic conditions in ethanol. The use of inert atmosphere to avoid a potential oxidation of o-phthalaldehyde leads to higher yields of both products from 17% to 25%. The usage of isopropanol instead of ethanol (synthesis of 2) significantly enhances yield from 25% to 55%.
[Cu(pap)Cl2] (1)
A solution of o-phthalaldehyde (67 mg, 0.50 mmol) and picolylamine (103 μl, 1.00 mmol) in 20 mL of ethanol (92%) was refluxed for 10 min, until its color changed to dark brown. Then CuCl2·2H2O (85 mg, 0.50 mmol) was added to the resulting mixture and an immediate change of color was observed with precipitation of a bright precipitate. This was dissolved under reflux and the dark clear solution was allowed to stand without disturbance for a week at 295 K. Thereafter the color of the solution changed to brown-green and dark green crystals were collected by filtration. These were suitable for X-ray diffraction analysis. Yield 38 mg (17%). Elem. anal.: calculated for [Cu(pap)Cl2]; 448.84 g mol−1; C20H18Cl2CuN4: C, 53.52; H, 4.04; N, 12.48%. Found: C, 53.05; H, 4.07; N, 12.22%; μeff = 1.7 BM.
[Co(pap)Cl2] (2)
Complex 2 was synthesized in a similar way as 1. The amounts of the reactants: o-phthalaldehyde (67 mg, 0.50 mmol), picolylamine (103 μl, 1.00 mmol), CoCl2·6H2O (117 mg, 0.500 mmol), 20 mL of ethanol (92%). The single crystals of 2 were grown after slow evaporation of the reaction mixture at 295 K over a time period of one week. Yield 38 mg (17%).
Elem. anal.: calculated for [Co(pap)Cl2]; 444.22 g mol−1; C20H18Cl2CoN4: C, 54.08; H, 4.08; N, 12.61%. Found: C, 53.86; H, 4.19; N, 12.02%; μeff = 4.3 BM.
{[Cu(H2O)(pap)]2(μ-ta)}·ta·8H2O (3)
An aqueous solution of 1 (45 mg, 0.10 mmol; 50 mL of water) was mixed with an aqueous transparent solution of dipotassium terephthalate {K2(ta)} in stoichiometric excess with respect to 1 (32 mg, 0.13 mmol; 10 mL of water). The mixture was placed in a round-bottom flask (100 mL volume) and refluxed for 45 minutes. A slight change of color was observed from green to bright green during the reaction. The hot solution was cooled slowly to room temperature and after two days a dark green crystalline precipitate was collected by vacuum filtration. The product loses a part of crystallization water molecules when dried in air what is accompanied with a slight change of color to bluish green. Yield 41 mg (58%). Elem. anal.: calculated for {[Cu(H2O)(pap)]2(μ-ta)}·ta·8H2O; 1264.20 g mol−1; C56H64Cu2N8O18: C, 53.29; H, 4.95; N, 8.88%. Found: C, 53.61; H, 4.48; N, 8.80%.
The crystals suitable for X-ray diffraction were grown by a slow diffusion method, starting from 1 mM aqueous solutions of the substrates and maintaining at least twofold stoichiometric excess of K2(ta). After two weeks of crystallization at 295 K, the crystals of sufficient quality were collected and the measurement of single crystal X-ray diffraction was carried in an Apiezon grease to avoid water loss. The identity of the single crystals with the bulk product (apart from the number of crystallization water molecules) was confirmed with elemental analyses and IR spectroscopy.
{[Co(H2O)(pap)]2(μ-ta)}·ta·4H2O (4)
The synthesis of 4 was carried out in a similar way as 3. Instead of 1, the complex 2 (44 mg, 0.10 mmol) was dissolved in 50 mL of water and mixed with 10 mL of aqueous solution of dipotassium terephthalate in stoichiometric excess (32 mg, 0.13 mmol). The mixture was refluxed for 45 minutes, and then it was allowed to cool at room temperature. After two days the red crystalline precipitate was collected by vacuum filtration. Yield 25 mg (42%). Elem. anal.: calculated for {[Co(H2O)(pap)]2(μ-ta)}·ta·4H2O; 1180.94 g mol−1; C56H54Co2N8O14: C, 56.95; H, 4.61; N, 9.49%. Found: C, 56.59; H, 4.74; N, 9.33%. The crystals suitable for X-ray diffraction were grown analogously as in the case of complex 3.
{[Co(pap)(suc)]·xH2O}n (5)
Complex 2 (95 mg, 0.21 mmol) was dissolved in 5 mL of water together with disodium succinate hexahydrate (63 mg, 0.23 mmol). The clear pale red solution was transferred to a 8 mL vial, sealed and thermostated at 373(1) K for 16 hours. During reaction time the solution colour turned dark red. The crystalline precipitate was filtered off and washed with water. Single crystals suitable for X-ray diffraction analysis were selected from the bulk. Yield 42 mg (37%). Elem. anal.: calculated for {[Co(pap)(suc)]·2.6H2O}n; 536.230 g mol−1; C24H27.2CoN4O6.6: C, 53.76; H, 5.11; N, 10.45%. Found: C, 53.70; H, 4.97; N, 10.43%; μeff = 3.8 BM.
Results and discussion
X-ray crystal structures
Mononuclear complexes. The X-ray molecular structures of 1 and 2, prepared in a one-pot reaction between o-phthalaldehyde, picolylamine and corresponding metal chlorides, are presented in Fig. 1. Crystallographic data, selected bond lengths and angles in 1 and 2 are listed in Tables 1 and 2. Both complexes are chiral and their divalent metal ions are coordinated by tridentate pincer type pap ligands forming five- and seven-membered chelate rings. Additional two cis-bound chlorido ligands neutralize the charge of the metal ions and make the complexes five-coordinate. The isolated complexes are the first examples of 1-ISND chelates and also rare examples of Cu(II) and Co(II) coordination compounds with seven-membered rings as evidenced by single-crystal X-ray diffraction.30–42 In both cases the conformations of the seven-membered rings may be described as boat-like.43 The bite angles of the ligands in five- and seven-membered rings are close to 80° and 100°, respectively for both complexes. The bulky pap ligands are puckered with N-picolyl part bent axially under angle 111° in both cases. The puckering of the iminoisoindoline, which may be described in terms of the dihedral angle between 1-picolyl and isoindoline planes is significantly different with values of 35.13° (for 1) and 16.46° (for 2). This difference is a consequence of disparate complex geometries adopted by the metal ions, despite possessing the same cis-[M(NNN)Cl2] coordination environment. In complex 1, copper(II) center coordinates one pap and two cis-chlorido ligands in a distorted square pyramidal (spy) manner whereas in complex 2 the coordination mode of cobalt(II) center may be described as a distorted trigonal bipyramid (tbpy). These findings are supported by several structural parameters. Firstly, in the case of 1, angles N2–Cu1–Cl2 (153.7°) and N4–Cu1–N1 (173.4°) give a trigonality degree (τ),44 equal to 0.3283 (for ideal spy geometry τ = 0). Analogously, in complex 2 angles N2–Co1–Cl2 (126.6°) and N4–Co1–N1 (177.8°) give τ = 0.8516 which is close to τ = 1, characteristic for the ideal trigonal bipyramidal geometry. Secondly, parameters which support the coordination mode assignment are the percentages of tetragonal elongation and trigonal compression of the M–Cl bonds.44 In the case of complex 1 the value of tetragonal elongation (9.639%) is strongly pronounced over trigonal compression (2.277%). On the other hand, in complex 2, these values are 0.08621% and 3.943%, respectively which is another evidence of trigonal bipyramidal coordination. Various coordination geometries of 1 and 2 are responsible for the significant differences in crystal packing between the complexes, as shown in Fig. 2 and S3 (ESI†). The key factor explaining this disparity is the interplay between weak hydrogen bonds in which chloride anions take part as acceptors and different π–π stacking patterns. Unlike for 1, the crystal data of 2 show the presence of cavities as well as the absence of ordered solvent molecules inside these cavities. The elemental analyses are also consistent with formulae containing fractional amounts of water (up to 0.5 molecules per Co). Similarly, the IR spectrum of 2 indicates the presence of water molecules by the appearance of O–H stretching bands at ca. 3440 cm−1. Interestingly, the crystal structure of 2 does not collapse without solvent molecules inside, the single crystal of 2 retains its crystallinity when dried and during crystal data collection at ambient conditions. Therefore, it seems that compound 2 contains water molecules that exhibit loose affinity to the hydrophobic surface of channels present in the structure of 2.
 |
| | Fig. 1 Molecular structures of 1 and 2 with atom labelling. Thermal ellipsoids represent 50% of displacement probability while the hydrogen atoms are shown as spheres of fixed radius. | |
Table 1 Crystallographic data for 1 and 2. Estimated standard deviations are shown in round brackets
| Compound |
1 |
2 |
| Parameters definition: R = Σ(|Fo| − |Fc|)/Σ(|Fo|); wR(F2) = {Σ[w(Fo2 − Fc2)2}1/2; w = 1/[σ2(Fo2) + (0.0808P)2 + 2.6927P]; P = (Fo2 + 2Fc2)/3. |
| Chemical formula |
C20H18Cl2CuN4 |
C20H18Cl2CoN4 |
| Formula weight |
448.82 |
444.21 |
| a [Å] |
8.221(5) |
8.733(5) |
| b [Å] |
14.202(5) |
12.221(5) |
| c [Å] |
16.421(5) |
13.101(5) |
| α, β, γ |
97.171(5)° |
64.207(5)°, 74.485(5)°, 85.967(5)° |
| μ [mm−1] |
1.44 |
7.66 |
| V [Å3] |
1902.2(15) |
1211.3(10) |
| Z |
4 |
2 |
| Space group |
P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| T [K] |
293 |
293 |
| λ [Å] |
0.71069 |
1.5418 |
| Dcalc [g cm−3] |
1.567 |
1.218 |
| R[F2 > 2σ(F2)]a |
0.053 |
0.043 |
| wR(F2)a |
0.160 |
0.116 |
Table 2 Selected bond lengths and angles for 1 and 2. Estimated standard deviations are shown in round brackets
| Bond |
Complex/bond length [Å] |
Angle |
Complex/[deg] |
| 1 |
2 |
1 |
2 |
| M1–Cl1 |
2.525(2) |
2.322(1) |
N1–M1–N4 |
173.4(1) |
177.8(1) |
| M1–Cl2 |
2.303(2) |
2.320(1) |
N4–M1–N2 |
96.4(1) |
99.7(1) |
| M1–N1 |
2.000(3) |
2.147(5) |
N2–M1–N1 |
79.7(1) |
78.0(1) |
| M1–N2 |
2.064(3) |
2.071(2) |
Cl2–M1–N2 |
153.7(1) |
126.64(8) |
| M1–N4 |
2.017(3) |
2.156(3) |
Cl1–M1–N2 |
95.75(9) |
108.69(8) |
| N2–C7 |
1.300(5) |
1.303(4) |
Cl1–M1–Cl2 |
110.08(5) |
122.74(4) |
| N3–C7 |
1.360(5) |
1.345(5) |
Cl1–M1–N4 |
93.5(1) |
93.2(6) |
| N3–C14 |
1.453(5) |
1.459(5) |
Cl1–M1–N1 |
92.2(3) |
87.4(1) |
 |
| | Fig. 2 Crystal-packing and intermolecular interactions in the lattice of 2 viewed along the c axis (A) and along the a axis (B). Packing along the a axis in 2 represented in a space fill model showing rectangular open channels (coloured in cyan and red) of 4.4 × 4.5 Å2 size, constituting 31% of unit cell volume (C). Hydrogen atoms are shown as spheres of fixed radius. | |
Dinuclear complexes. The reactions of 1 and 2 with dipotassium terephthalate in aqueous solutions lead to crystalline solids of 3 and 4, respectively. These compounds were found to be iso-structural and their molecular structures are presented in Fig. 3 (3) and S4† (4). The selected crystallographic data, bond lengths and angles for the two complexes are given in Tables S1† and 3.
 |
| | Fig. 3 Molecular structure of {[Cu(H2O)(pap)]2(μ-ta)} (3) with the atom labelling scheme and 50% thermal displacement ellipsoids. Hydrogen atoms are shown as spheres of fixed radius whereas crystallization water molecules and non-coordinated terephthalate dianion were omitted for clarity. | |
Table 3 Selected bond lengths and angles for 3 and 4. Estimated standard deviations are shown in round brackets
| Bond |
Bond length [Å] |
Angle |
Complex/[deg] |
| 3 |
4 |
3 |
4 |
| M1–O1 |
2.255(2) |
2.037(2) |
N1–M1–N2 |
82.28(6) |
80.11(5) |
| M1–N1 |
2.008(2) |
2.120(2) |
N2–M1–N4 |
99.43(6) |
100.99(5) |
| M1–N2 |
2.016(1) |
2.078(1) |
O2–M1–O3 |
53.32(4) |
58.41(4) |
| M1–N4 |
2.024(1) |
2.136(1) |
C5–C6–N2 |
110.7(1) |
112.2(1) |
| M1–O2 |
2.0021(8) |
2.1279(9) |
C6–N2–C7 |
117.3(1) |
116.3(1) |
| M1–O3 |
2.737(1) |
2.361(1) |
N2–C7–N3 |
122.4(2) |
122.3(1) |
| C6–N2 |
1.467(2) |
1.472(2) |
N2–C7–C8 |
131.4(2) |
131.8(1) |
| N2–C7 |
1.305(2) |
1.308(2) |
C7–N3–C15 |
125.2(1) |
125.4(1) |
| C7–N3 |
1.358(2) |
1.361(2) |
C7–N3–C14 |
114.0(1) |
114.1(1) |
| N3–C14 |
1.458(2) |
1.455(2) |
N3–C15–C16 |
110.1(1) |
111.3(1) |
Each cobalt(II) or copper(II) center exhibits a distorted octahedral geometry with a terminally bound tridentate pap ligand and a water molecule, as well as a bridging terephthalate in the μ-η2 coordination mode. The carboxylate groups of the bridging terephthalate adopt highly asymmetrical arrangements around metal centers (with M–O bond lengths 2.002 and 2.737 Å for Cu, and 2.128 and 2.361 Å for Co). The seven-membered ring conformations of the pap ligand are similar to those of 1 and 2, however, unlike in 1, the iminoisoindoline moieties are almost perfectly planar. Interestingly, the terephthalate dianion, except acting as the bridging ligand, occupies additional position in the crystal lattice of the binuclear complexes. It is strongly hydrogen-bonded (Fig. 4 and S4†) with the coordinated aqua ligands (D⋯A distance is 2.7 Å for 3 and 2.6 Å for 4) as well as crystallization water molecules. It acts as a cross-linker between adjacent binuclear complexes and it also compensates their positive charge. The binuclear units are stacked together by π–π interactions between planar iminoisoindoline moieties. All these supramolecular interactions lead to a formation of channels (approx. 2 × 8 Å2 for both complexes) along the a axis (Fig. 4), occupied by water molecules. Upon dehydration, carried out at conditions indicated by TGA curves (Fig. S7, ESI†), the compounds 3 and 4 change irreversibly as evidenced by PXRD patterns, ATR-FTIR and diffuse reflectance UV-VIS spectra (Fig. S8, ESI†).
 |
| | Fig. 4 Crystal structure of 4 viewed along the a axis (H atoms have been omitted for clarity). Various types of non-covalent interactions are indicated (water molecules have been omitted). Co, O, N, and C atoms are shown in purple, dark grey, blue, and grey, respectively. | |
Polynuclear complex. The reaction of 2 with disodium succinate results in formation of polymeric complex {[Co(pap)(suc)]·2.6H2O}n (5) whose molecular structure and atom labelling are presented in Fig. 5. The crystallographic data for 5 are given in Table S2† together with comments on the refinement and disorder rationalisation. The selected bond lengths and angles are included in Table 4. The succinate moiety and crystallization water molecules are disordered what was not shown in the figures.
 |
| | Fig. 5 (A) Molecular structure of 5 together with atom labelling. Thermal ellipsoids represent 50% of displacement probability. Hydrogen atoms as well as the set B of disordered succinate atoms were not shown for clarity. Symmetry code for primed atoms: −x, −½ + y, ½ − z. (B) The 2D ball and stick arrangement of zigzag chains (green color represent the upside plane while magenta color represent the lower plane in relation to direction perpendicular to bc plane) through two types of π–π stacking interactions: between isoindoline phenyl rings and 1-imine bond and pirydyl (N1) ring (blue dashed lines). The crystallization water molecules were omitted for clarity. | |
Table 4 Selected bond lengths and angles for 5. Estimated standard deviations are shown in round brackets
| Bonds [Å] |
Angles [deg] |
| Co1–O1A |
2.287(4) |
N1–Co1–N2 |
79.88(9) |
| Co1–N1 |
2.113(2) |
N2–Co1–N4 |
101.6(1) |
| Co1–N2 |
2.082(2) |
O2A–Co1–O3A |
108.0(1) |
| Co1–N4 |
2.115(3) |
C5–C6–N2 |
111.8(2) |
| Co1–O2A |
2.286(3) |
C6–N2–C7 |
116.3(2) |
| Co1–O3A |
2.152(4) |
N2–C7–N3 |
122.1(2) |
| C6–N2 |
1.467(4) |
N2–C7–C8 |
131.7(2) |
| N2–C7 |
1.311(3) |
C7–N3–C15 |
125.8(2) |
| C7–N3 |
1.361(4) |
C7–N3–C14 |
113.4(2) |
| N3–C14 |
1.455(4) |
N3–C15–C16 |
112.1(3) |
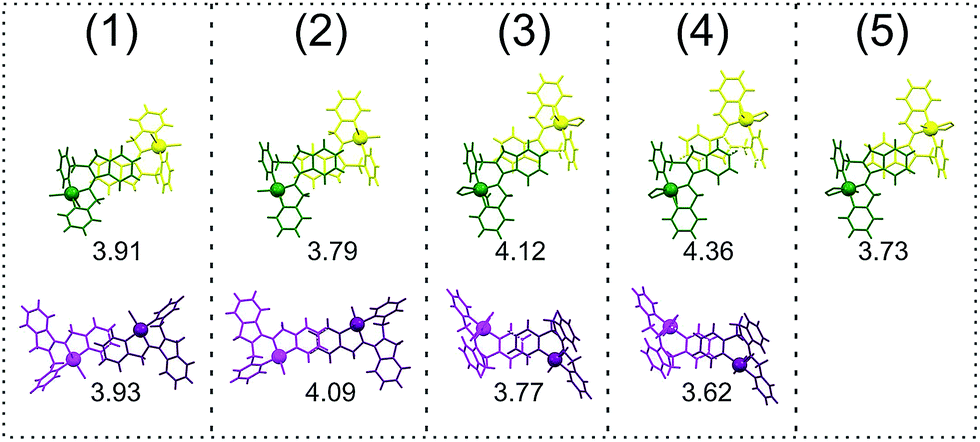
The cobalt center in 5 adapts distorted octahedral geometry with pap ligand coordinated meridionally through N1, N2 and N4 nitrogen atoms. The rest of three coordination places are occupied with oxygen donor atoms from disordered succinate dianion. Interestingly, the dicarboxylate linker is coordinated to metal center in a symmetrically chelating mode, by O1 and O2 oxygen atoms, as well as in a monodentate manner, by O3 atom (μ-η2,η1). The O4 oxygen atom remains uncoordinated and hydrogen bonded with the only one ordered water molecule in the crystal lattice. The cobalt–oxygen bond lengths are slightly longer for chelating oxygen atoms due to steric reasons. On the other hand, the axial cobalt–nitrogen bonds (N1–Co1 and N4–Co1) are slightly shorter than equatorial bond (N2–Co1). The bite angles of the pap ligand are almost identical as previously described (1–4). The diversified coordination manner of succinate dianion, stemming from its conformational flexibility, results in the formation of a 1D zigzag chain polymer as shown in Fig. 5B. The chains are arranged in a 3D supramolecular framework through various π–π stacking interactions. However, in contrast to 3 and 4, the crystal packing in 5 does not facilitate occurrence of large voids due to compact size of succinate linker. Two types of π–π stacking interactions can be found in the crystal structure of 5. These types of interactions have been compared for all complexes 1–5 in Fig. 6. In case of the complexes 1–4 each structure contains three types of π–π stacking interactions with various centroid–centroid distances being influenced not only by steric crowding of bulky pap ligand and coligands but predominantly by dissimilar coordination geometries imposed by different metal centers. The strongest interaction between phenyl rings, in terms of centroid–centroid distance, of isoindoline parts of pap ligand occurs in the structure of polymeric 5. In crystal lattices of 3 and 4, this interaction has rather C–H⋯π character, due to large interplanar separation exceeding 4 Å. The π–π interaction between pirydyl rings containing N4 nitrogen atom can be observed only for complexes 1–4. The strongest of these occurs in the structure of 4, while in case of 2 it has rather C–H⋯π character than π–π, with centroid–centroid distance above 4 Å. The closest intermetallic non-bonding distance in the structure of 5 is 6.6022(9) Å, similarly as in the crystal structure of 4 (6.5948(4) Å). Contrary to dimers 3 and 4, compound 5 may be reversibly dehydrated (Fig. S8†) and is thermally stable up to 570 K (Fig. S7†). TG curve of the as-synthesized 5 reveals that, when heated, it loses five water molecules per Co whereas elemental analysis findings support the crystal data for air-dried 5 indicating fractional amount of 2.6 H2O molecules per cobalt centre. One of them (O6) is disordered and fractionally occupies three various sites.
 |
| | Fig. 6 The comparison of π–π stacking interactions in crystal structures of 1–5 in capped sticks projection (the metal atoms are represented as spheres of arbitrary radii). The numbers refer to centroid–centroid distances in Å. The rows top to bottom correspond to interactions: (i) phenyl rings of isoindoline; (ii) pirydyl rings (containing N4). | |
IR spectroscopy
The infrared spectroscopic data of both complexes 1 and 2 exhibit several characteristic absorptions for the pap organic ligand (Fig. S5 and S6†).45 The ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) absorptions appear as strong bands at 1620 and 1606 (1) as well as at 1602 with a shoulder at ca. 1620 cm−1 (2). Ring-breathing vibrations of the aromatic rings of the organic ligand range from 1420 to 1585 cm−1.
N) absorptions appear as strong bands at 1620 and 1606 (1) as well as at 1602 with a shoulder at ca. 1620 cm−1 (2). Ring-breathing vibrations of the aromatic rings of the organic ligand range from 1420 to 1585 cm−1.
The IR spectra of 3 and 4 (Fig. S5†) show additionally bands characteristic for a carboxylate group. The ν(CN) absorptions appear as strong bands at 1609 and 1597 (3) as well as at 1604 and 1597 cm−1 (4), all wavenumbers slightly lower than those of corresponding mononuclear complexes (1 and 2). The carboxylate bands are readily identified in the spectra as distinctive bands, not observed for 1 and 2. Strong absorptions at 1563, 1570 cm−1 (for 3) and at 1567 cm−1 (for 4) were assigned to νas(COO), whereas those at 1359, 1396 cm−1 (for 3) and at 1364, 1403 cm−1 (for 4) were assigned to νs(COO) in μ-η2 carboxylate groups. The IR spectrum of 5 is presented in Fig. S6.† Similarly as in the spectra of 1–4 discussed above, very strong overlapping bands are present in the range 1520–1600 cm−1. In this range the azomethine stretching bands as well as pirydyl ring breathing vibrations can be found together with asymmetric stretching of the succinate carboxylate group coordinated in a monodentate manner to cobalt center. The medium to strong overlapping bands present in the range 1520–1300 cm−1 may be ascribed to symmetric stretching of succinate carboxylate groups and pirydyl ring breathing vibrations.
UV-VIS spectroscopy
The diffuse reflectance spectra of 1–5 together with excitation and emission spectra in the solid state are presented in Fig. 7. The reflectance spectra of the complexes, beside peaks at ca. 250 nm, contain numerous shoulders in the UV region, which may be ascribed to intraligand π → π* transitions in the pap entity. In the visible range, weak n → π* transitions in the azomethine group of 1-iminoisoindoline may be distinguished as shoulders at ca. 400 nm.46 In the region above 400 nm several overlapping bands for 1–5 are observed and their unambiguous assignments as those of CT or d–d origin, are difficult without supportive quantum chemical calculations.
 |
| | Fig. 7 The UV-VIS reflectance spectra for 1–5 at room temperature (left side, black solid line). The luminescence spectra for solids 1–5 at room temperature (right side). The blue dashed line denotes excitation spectra (registered for emission at 600 nm) while the red solid line denotes emission spectra (excitation at 400 nm). | |
In each emission spectrum of complexes 1–5, two bands of highest intensity appear at the same position at ca. 520 and 600 nm. This clearly indicates that luminoforic processes involve intraligand transitions within the pap pincer, present in all compounds.
Conclusions
In summary, we have demonstrated the first syntheses and characterization of cobalt(II) and copper(II) complexes with an in situ prepared novel asymmetric trinitrogen isoindoline-based pincer type ligand. Both complexes consist of (a) stable, luminescent, hydrophobic part including the tridentate pincer ligand and the metal center; (b) labile hydrophilic part with cis-dichlorido coligands; and (c) paramagnetic metal centers. In this work we have shown that these compounds may be successfully used as molecular building blocks for the synthesis of three new hydrated supramolecular networks. The formation of these networks is accompanied by the substitution of the labile chloride ligands and the retention of the pincer ligand with luminescent property. We envisage that the monomeric complexes may have wider applicability, as molecular tectons, in the construction of either discrete polynuclear clusters or extended networks with desirable properties. Upon labile chlorides dissociation, the complexes become cationic and are likely to strongly interact with such moieties as e.g. ionizable organic ligands or anionic metalloligands. Moreover, the meridionally coordinated isoindoline derivative, acting as an inert blocking ligand, together with labile coligands, creates opportunities for the use of the complexes in catalysis.
Acknowledgements
The authors gratefully acknowledge National Science Centre in Poland (grant no. 2012/07/B/ST5/00904) for the financial support of this research. The research was carried out partially with the equipment purchased thanks to the financial support of the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (contract no. POIG.02.01.00-12-023/08).
Notes and references
- C. J. Moulton and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1976, 1020–1024 RSC.
- G. van Koten and R. J. M. (Bert) Klein Gebbink, Dalton Trans., 2011, 40, 8731–8732 RSC.
- G. van Koten, Pure Appl. Chem., 1989, 61, 1681–1694 CrossRef CAS.
- M. Albrecht and M. M. Lindner, Dalton Trans., 2011, 40, 8733–8744 RSC.
- V. A. Kozlov, D. V. Aleksanyan, M. V. Korobov, N. V. Avramenko, R. R. Aysin, O. A. Maloshitskaya, A. S. Korlyukov and I. L. Odinets, Dalton Trans., 2011, 40, 8768–8772 RSC.
- E. M. Schuster, M. Botoshansky and M. Gandelman, Dalton Trans., 2011, 40, 8764–8767 RSC.
- K. Yoshida, T. Nakashima, S. Yamaguchi, A. Osuka and H. Shinokubo, Dalton Trans., 2011, 40, 8773–8775 RSC.
- G. Hamasaka, T. Muto and Y. Uozumi, Dalton Trans., 2011, 40, 8859–8868 RSC.
- M. H. P. Rietveld, D. M. Grove and G. van Koten, New J. Chem., 1997, 21, 751–771 CAS.
- S. Wanniarachchi, B. J. Liddle, J. Toussaint, S. V. Lindeman, B. Bennett and J. R. Gardinier, Dalton Trans., 2011, 40, 8776–8787 RSC.
- R. A. Gossage, Dalton Trans., 2011, 40, 8755–8759 RSC.
- R. B. Lansing, K. I. Goldberg and R. A. Kemp, Dalton Trans., 2011, 40, 8950–8958 RSC.
- J. L. Cryder, A. J. Killgore, C. Moore, J. A. Golen, A. L. Rheingold and C. J. A. Daley, Dalton Trans., 2010, 39, 10671–10677 RSC.
- B. F. Wicker, M. Pink and D. J. Mindiola, Dalton Trans., 2011, 40, 9020–9025 RSC.
- P. Zuman, N. Salem and E. Kulla, Electroanalysis, 2009, 21, 645–649 CrossRef CAS.
- Y. Mulyana, C. J. Kepert, L. F. Lindoy and J. C. McMurtrie, Eur. J. Inorg. Chem., 2005, 12, 2470–2475 CrossRef.
- L.-N. Zhu, S. Gao and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2618 CAS.
- Z. Deng, L. Huo, L. Zhu, H. Zhao and S. Gao, Polyhedron, 2010, 29, 3207–3213 CrossRef CAS PubMed.
- J. M. Chitanda, D. E. Prokopchuk, J. Wilson Quail and S. R. Foley, Organometallics, 2008, 27, 2337–2345 CrossRef CAS.
- J. M. Chitanda, S. Wu, J. W. Quail and S. R. Foley, Inorg. Chim. Acta, 2011, 379, 122–129 CrossRef CAS PubMed.
- J. M. Chitanda, D. E. Prokopchuk, J. W. Quail and S. R. Foley, Dalton Trans., 2008, 6023–6029 RSC.
- J. M. Chitanda, J. Wilson Quail and S. R. Foley, J. Organomet. Chem., 2009, 694, 1542–1548 CrossRef CAS PubMed.
- CrysAlis PRO, Agilent Technologies, Oxford, United Kingdom, 2010 Search PubMed.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 2007, 64, 112–122 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 2002, 58, 389–397 CrossRef PubMed.
- S. Wolff, D. Grimwood, J. McKinnon, M. Turner, D. Jayatilaka and M. Spackman, CrystalExplorer 3.0, University of Western Australia, Perth, Australia, 2012 Search PubMed.
- S. Hamamci, V. T. Yilmaz and W. T. Harrison, J. Coord. Chem., 2003, 56, 1033–1039 CrossRef CAS.
- G. Minardi, E. Mura, A. M. Pistuddi, C. Solinas, A. Bacchi, C. Pelizzi, G. Pelizzi and G. Chelucci, Transition Met. Chem., 1999, 24, 481–485 CrossRef CAS.
- K. Ida, H. Sakiyama, H. Ōkawa, N. Matsumoto, Y. Aratake, I. Murase and S. Kida, Polyhedron, 1992, 11, 65–70 CrossRef CAS.
- M. M. Kadooka, L. G. Warner and K. Seff, Inorg. Chem., 1976, 15, 812–816 CrossRef CAS.
- Z. Puterová, J. Valentová, Z. Bojková, J. Kožíšek and F. Devínsky, Dalton Trans., 2011, 40, 1484–1490 RSC.
- H.-B. Zhu, Z.-Y. Dai, W. Huang, K. Cui, S.-H. Gou and C.-J. Zhu, Polyhedron, 2004, 23, 1131–1137 CrossRef CAS PubMed.
- R. Balamurugan, M. Palaniandavar and R. S. Gopalan, Inorg. Chem., 2001, 40, 2246–2255 CrossRef CAS PubMed.
- C. Richardson and P. J. Steel, Aust. J. Chem., 2000, 53, 93–97 CrossRef CAS.
- V. Broughton, G. Bernardinelli and A. F. Williams, Inorg. Chim. Acta, 1998, 275, 279–288 CrossRef.
- I. Burshtein, V. Pavlovskii and A. Poznyak, Russ. J. Coord. Chem., 1998, 24, 131–135 CAS.
- H. Maluszyńska, A. Perkowska and E. Skrzypczak-Jankun, J. Chem. Crystallogr., 1995, 25, 19–23 CrossRef.
- J. W. Martin, J. H. Timmons, A. E. Martell, P. Rudolf and A. Clearfield, Inorg. Chem., 1981, 20, 814–821 CrossRef CAS.
- C. Prout, J. Carruthers and F. Rossotti, J. Chem. Soc. A, 1971, 3342–3349 RSC.
- R. Kadyrov, A. Börner and R. Selke, Eur. J. Inorg. Chem., 1999, 705–711 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- G. Socrates, Infrared and Raman characteristic group frequencies: tables and charts, Wiley, Chichester, 3rd edn, 2001 Search PubMed.
- I. Sović, V. Stilinović, B. Kaitner, S. Kraljević-Pavelić, M. Bujak, K. Čuljak, P. Novak and G. Karminski-Zamola, J. Mol. Struct., 2011, 1006, 259–265 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Graph-set analysis of hydrogen bonding of 1 and 2, comment on the refinement of 5, ATR-FTIR spectra for 1–5, TG and PXRD data for 3–5, molecular structure of 4, crystal packing in 1, conductivity measurements for 1 and 2 in aqueous solutions. CCDC 974887, 974888, 980973, 980974 and 1023714. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra02063h |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.