
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theoretical investigations of novel zinc oxide polytypes and in-depth study of their electronic properties†
D.
Zagorac
*ab,
J. C.
Schön
a,
J.
Zagorac
ab and
M.
Jansen
a
aMax Planck Institute for Solid State Research, Stuttgart, Germany
bInstitute of Nuclear Sciences Vinča, Materials Science Laboratory, University of Belgrade, Belgrade, Serbia. E-mail: dzagorac@vinca.rs
First published on 4th March 2015
Abstract
Zinc oxide is one of the most investigated compounds in materials science, both experimentally and theoretically, while in nature it appears only rarely, as the mineral zincite. Yet there are still many open questions: Is it still possible to observe or synthesize new modifications of zinc oxide? And can we improve the properties of a material that has already been investigated in thousands of studies? What is the connection between zincite, zinc sulfide and zinc oxide, and can we finally explain the controversial mineral matraite? In short, Yes: the answer to these questions is polytypism. We identify a multitude of possible stable polytypes for zinc oxide, and we show that by varying the stacking order, we can fine-tune the electronic properties such as the direct primary and secondary band gaps in zinc oxide without adding dopant atoms.
1. Introduction
In the literature, polytypism describes the phenomenon where a chemical compound crystallizes in a variety of periodically repeated layered structures – the polytypes.1 Usually, the individual layers constituting the polytypes are structurally identical, however the position of these layers relative to their neighboring layers can vary, leading to a multitude of stacking variants. Although quite a number of examples of compounds crystallizing in two or three such polytypes exist,2–6 it is not as common a phenomenon in experimental solid state chemistry as one would expect. There are still many open questions, e.g. why do some compounds show polytypism and others do not, how many polytypes can exist of one compound, and what is the effect of polytypism on the mechanical, thermal, or electronic properties?One of the compounds where many polytypes are expected to exist is zinc oxide (ZnO), since the closely related zinc sulfide (ZnS) exhibits about 200 experimentally identified stacking variants.2 However, only three bulk phases of ZnO are known experimentally, a wurtzite and a sphalerite modification at ambient conditions, and a rocksalt phase at high pressures.7 As ZnO is a technologically very important compound, new modifications with different, possibly tunable properties would be highly desirable. Preliminary theoretical studies8 and subsequent work on nanofilms9 suggest that such polytypes should be capable of existence. Using global energy landscape exploration techniques, we have now identified a multitude of stable stacking variants of the wurtzite and the sphalerite type structure in zinc oxide, which are energetically nearly as low as the global minimum, the wurtzite modification.
In this study, we present the results of these global searches on the energy landscape of ZnO and analyze this plethora of newly discovered zinc oxide polytypes. We show that by varying the stacking order, we can fine-tune electronic properties such as the direct primary and secondary band gaps in zinc oxide.
Quite generally, all stable and metastable modifications of a chemical system correspond to the locally ergodic regions on the energy/enthalpy landscape of the system. At low or medium temperatures, such regions consist of local minima surrounded by sufficiently high energy barriers.10,11 As described in detail in the methods section, we proceeded in two steps during the global exploration of the energy landscape of zinc oxide: first, a number of local minima were identified using simulated annealing as a global optimization procedure,8,12 and in a second step, these minima served as starting points for further global explorations using the threshold algorithm.13,14 In the latter method, one performs random walks, usually starting from a local minimum, where every move is accepted as long as the new configuration remains below a given sequence of energy lids. Periodically, the random walker is quenched into the nearest local minimum, in order to find new local minima and to estimate the barriers separating them. Since we are interested in modifications that are stable at ambient conditions, we set the pressure to p = 0.1 MPa for both the simulated annealing and the threshold runs (for simulated annealing based explorations of the enthalpy landscape of ZnO over a wide range of pressures, cf.ref. 12).
2. Computational methods
Our general approach to the determination of structure candidates is based on the search for local minima and proceeds via the global exploration of the energy landscape of the system of interest. The method has been given in detail elsewhere;10,15 here, we just provide information specific to this study. The minima were identified using simulated annealing as a global optimization procedure.16 Since the global search involved many millions of energy evaluations, we employed an empirical energy/enthalpy function, E = Epot + pV, in this study. Here, Epot = Σ〈i,j〉Vij(rij), where Vij is a two-body empirical potential consisting of Lennard-Jones and Coulomb terms: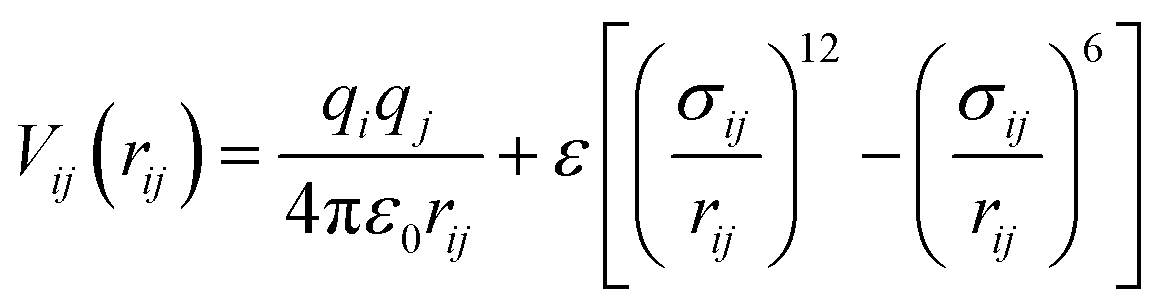 | (1) |
Here, qi and qj are the charges of the ions, rij is the distance between atoms i and j, σij = rs(i)rion(i) + rs(j)rion(j) is the scaled sum of the ionic radii, and ε gives the strength of the Lennard-Jones potential.12 For the evaluation of the Coulomb summation, the method proposed by DeLeeuw was employed.17 Regarding the number of atoms in the simulation cell, we performed the global optimizations for 1, 2, 3, 4 and 5 formula units ZnO per simulation cell at standard pressure. Both the atom positions (70% of all Monte Carlo steps move an individual atom, 10% swap the positions of two atoms) and the parameters of the periodically repeated simulation cell (20% of all Monte Carlo steps) were freely varied during the random walks. Each simulated annealing run consisted of 4 × 106 Monte Carlo steps, followed by 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 stochastic quench steps.
000 stochastic quench steps.
The barrier structure is explored using the threshold algorithm,14 where the landscape accessible from a local minimum below a sequence of energy barriers (thresholds) is systematically explored for all important local minima. In this study, we have again employed the two-body empirical potential consisting of Lennard-Jones and Coulomb terms, using up to 6 formula units. During these calculations the number of formula units Z and the initial structure were varied. Each calculation consisted of 28 lids, ranging from −6.3 to −3.9 eV, with a step size of 0.1 eV. In both cases, for the global exploration and the threshold algorithm calculations, we have used the modular G-42 code.10
Subsequent to the global search, the candidates found were locally optimized on ab initio level, and the E(V) and H(p) curves for the various modifications were computed, in order to be able to determine the thermodynamically stable ones as function of pressure. For this, we employed the program CRYSTAL09,18 based on linear combination of atomic orbitals (LCAO), and for the local optimizations we employed analytical gradients.19 Each local optimization was performed on the Hartree–Fock and the density functional theory (DFT) level, using the local density approximation (LDA). In addition we have employed a hybrid B3LYP functional (Becke's three-parameter functional in combination with the correlation functional of Lee, Yang, and Parr20). It is reasonable to use several different ab initio methods, in order to get some feeling for the quantitative validity of the results.21,22
For these local optimizations, all electron basis sets (AEBS) based on Gaussian-type orbitals (GTO) were used. In the case of Zn2+ a [6s5p2d] basis set was used as in ref. 23 and 24. For O2− a [4s3p] basis set was used as in ref. 25 and 26 (see also ESI†). During local optimizations a k-point sampling net of size 8 × 8 × 8 was used. In addition, a smearing temperature of 0.01Eh was applied during the local optimization and the calculation of the E(V) and H(p) curves, in order to facilitate the numerical integration. For the analysis of the structures and their visualization we used KPLOT27 and the VESTA28 program.
3. Results
The global searches at standard pressure resulted in a plethora of local minima, which belong to a variety of structure types, such as the wurtzite, sphalerite, 5–5, and β-BeO type (at high pressures, additional modifications were found, exhibiting e.g. the NaCl, the NiAs, the GeP or the CsCl type12). The appearance of these structure types was not surprising, since they also have been found on the energy landscape of a large number of other AB-systems such as the earth alkaline oxides29 or the alkali halides.30However, during the threshold runs several modifications with very low energies comparable to the global minimum were found. These modifications correspond to stacking variants of the wurtzite and sphalerite structures, i.e., these modifications are the missing polytypes of zinc oxide (see Fig. 1). In order to confirm that these polytypes are not artefacts of the potential used, further simulated annealing runs using a Born–Mayer-type potential (eqn (1)) were performed, yielding again these polytypes as local minima on the landscape. Furthermore, we note that when we have chosen one of the polytypes as starting minimum for the threshold runs, we required lid values of at least 0.5 eV per atom, in order to leave this minimum basin on the energy landscape. Finally, the same periodic modification was observed with many different types of unit cells (while one often prefers to employ symmetry adapted unit cells for a structure description, there exist an infinite number of compatible unit cell of various sizes that can describe the same infinite periodic structure, many of which appear in landscape explorations when the simulation cell can be varied), again underlining the fact that these polytypes correspond to stable states on the energy landscape of ZnO.
 | ||
| Fig. 1 Visualization of the experimentally observed (a and b) and calculated (c–i) modifications of zinc oxide: (a) wurtzite (2H) type; (b) sphalerite (3C) type; (c) 4H polytype; (d) 5H polytype; (e) 6H polytype; (f) 8H polytype; (g) 9R polytype; (h) 12R polytype; (i) 15R polytype. | ||
Regarding the ab initio level, the local minimizations were performed both with and without symmetry prescriptions during the gradient-based minimization. Furthermore, we have changed the cell parameters and displaced the atoms from their apparent equilibrium values by finite amounts for all the minimum configurations, and repeated the local minimization procedure with three different ab initio energy functions. In all cases, the structure returned to the stable position obtained originally. We note that this was true for both those polytypes that had appeared during the simulated annealing and threshold runs (4H, 5H, 6H, 9R) and those that had been constructed in analogy to the ZnS-system (8H, 12R, 15R).
3.1 Energetic ranking and structural features of the new polytypes
The local optimizations of all the stable structure candidates on ab initio level show that the wurtzite and sphalerite polymorphs of zinc oxide are the energetically lowest and thermodynamically most stable ones, which is in agreement with experimental observations7,31–34 and previous calculations.35–41 The wurtzite type together with the sphalerite type (see Fig. 1a) are the stable modifications at ambient conditions. For calculations performed on LDA and B3LYP level, these two minima have essentially the same energy, while on the Hartree–Fock level, their energies differ, E(wurtzite) = −1852.7170Eh < E(sphalerite) = −1852.7143Eh (see Table 1). Both modifications are well known in the zinc/iron sulfides with the analogous composition, (Zn, Fe)S. Wurtzite (B4) exhibits space group P63mc and its structure is composed of ZnO4 tetrahedra that are stacked in a hexagonal sequence ABABAB… (hcp-packing). Sphalerite (B3) exhibits space group F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m, and its structure can be visualized as an ABCABCABC… cubic sequence of ZnO4 tetrahedra (fcc-packing).8
3m, and its structure can be visualized as an ABCABCABC… cubic sequence of ZnO4 tetrahedra (fcc-packing).8
| Structure type | Energy (Eh) | ||
|---|---|---|---|
| LDA | B3LYP | HF | |
| Wurtzite (2H) | −1851.2629 | −1854.5584 | −1852.7170 |
| (0 K) | (0 K) | (0 K) | |
| 4H | −1851.2625 | −1854.5582 | −1852.7150 |
| (+125 K) | (+60 K) | (+630 K) | |
| 5H | −1851.2630 | −1854.5581 | −1852.7159 |
| (−30 K) | (+90 K) | (+350 K) | |
| 6H | −1851.2631 | −1854.5580 | −1852.7157 |
| (−60 K) | (+125 K) | (+410 K) | |
| 8H | −1851.2630 | −1854.5579 | −1852.7146 |
| (−30 K) | (+160 K) | (+760 K) | |
| 9R | −1851.2585 | −1854.5552 | −1852.7165 |
| (+1400 K) | (+1000 K) | (+160 K) | |
| 12R | −1851.2627 | −1854.5578 | −1852.7153 |
| (+60 K) | (+190 K) | (+540 K) | |
| 15R | −1851.2513 | −1854.5509 | −1852.7150 |
| (+3660 K) | (+2700 K) | (+630 K) | |
| Sphalerite (3C) | −1851.2632 | −1854.5575 | −1852.7143 |
| (−95 K) | (+285 K) | (+850 K) | |
The appearance of the 4H, 5H, 6H, and 9R stacking polytypes (see Fig. 1 and 2) in the global search and threshold runs is a clear indication that many other polytypes might be capable of existence in the ZnO system. The symbols used to classify the ZnO polytype structures have a specific meaning; e.g. the number 3 in the 3C polytype refers to the three layer periodicity of the stacking (ABC) and the letter C denotes the cubic symmetry of the crystal. The wurtzite ABAB… stacking sequence is denoted as 2H, where 2 stands for two layer stacking periodicity and H for hexagonal symmetry. This periodicity doubles and triples in 4H and 6H polytypes. The family of rhombohedral polytypes is labelled with R, for example 9R2 (see Fig. 1). Therefore, we performed a series of local optimizations on ab initio level starting from other polytype structures known from ZnS: 8H, 12R and 15R.2,42 Of course, there exist many more hypothetical polytypes of ZnS and their stacking variants classified via the Zhdanov symbol; however, since their unit cells increase with the polytype number, we have not included them into the local ab initio energy minimizations. We would like to note a difference to the corresponding ZnS system, since there exist no experimental or theoretical data of a 5H polytype in the ZnS system. On the other hand, the 5H polytype structure of ZnO that we have found in our global search and threshold runs, and that we have later optimized on ab intio level, shows very good agreement with the 5H polytype structure from the SiC system.43
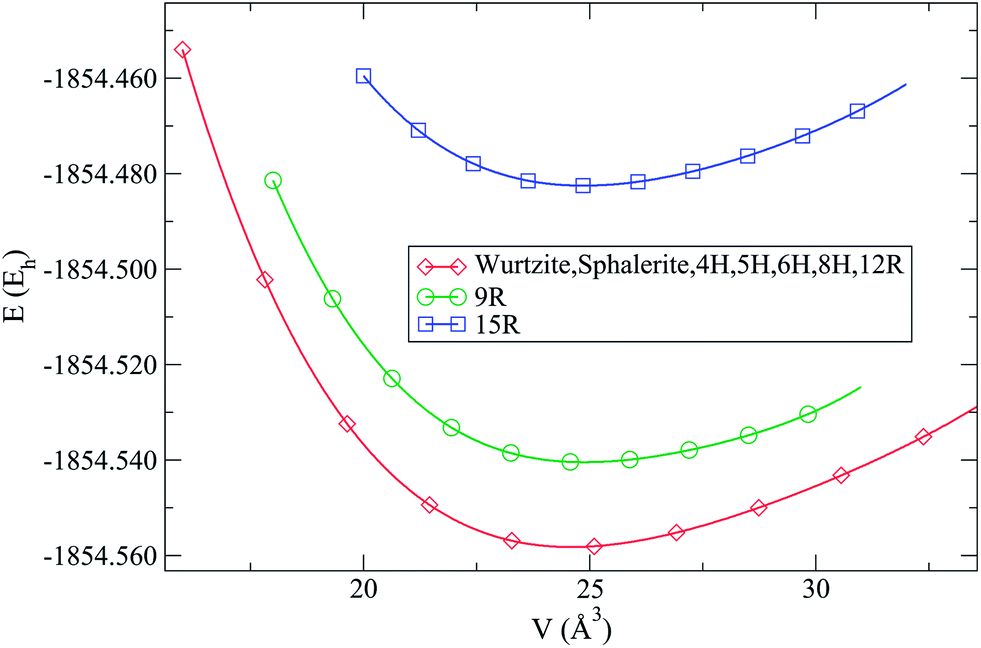 | ||
| Fig. 2 Calculated E(V) curves for the new stacking variants of ZnO at B3LYP level. Energies per formula unit are given in hartrees (Eh). Note that most of the calculated ZnO polytypes (4H, 5H, 6H, 8H and 12R), as well as sphalerite structure, exhibit essentially the same E(V) curve as wurtzite modification at this level of calculation and are indistinguishable on the scale of the figure, and therefore only the curve for wurtzite has been plotted. | ||
Using DFT (LDA and B3LYP), we observe that several polytypes (4H, 5H, 6H, 8H and 12R) have nearly the same energy as the wurtzite (2H) and sphalerite (3C) modifications (see Fig. 2 and Table 1). Two polytypes were higher in energy at this level of calculations: the 9R and the 15R types. The 9R polytype was only slightly higher in energy at e.g. the B3LYP level (E(9R) − E(2H) = 0.0032Eh, corresponding to about 1000 K), but the 15R stacking variant could only be optimized as a distorted version, and was rather high in energy on DFT level.
Using Hartree–Fock (see Table 1), we observe that the 12R polytype is somewhat higher in energy, while the 9R polytype has an energy similar to the one of the wurtzite-type. Furthermore, the distorted 15R polytype, and also the 4H, 5H, 6H and 8H variants, are similar in energy to the sphalerite-type modification (see ESI†). A summary of structural data for each of the calculated ZnO polytypes including the full Zhdanov symbol is presented in the ESI.†
3.2 Electronic properties of the polytypes
For the new predicted ZnO polytypes, we have performed band structure and density of states (DOS) calculations, in order to gain additional insight into the electronic properties of these possible modifications of zinc oxide. The calculations were performed using Hartree–Fock, DFT (LDA) and hybrid (B3LYP) functionals. Our calculations were in good agreement with previous experimental7,34,44,45 and theoretical findings,35,46–51 for those modifications where such data were available. Table 2 shows the band gaps calculated using different functionals for each of the ZnO polytypes investigated.| Structure type | Primary direct band gap (eV) | Secondary direct band gap (eV) | ||||
|---|---|---|---|---|---|---|
| LDA | B3LYP | HF | LDA | B3LYP | HF | |
| a The rhombohedral unit cell has been converted to a hexagonal unit cell for comparison. The secondary band gap occurs along the H–K direction of the Brillouin zone, instead of at the A point, as in those polytypes that are naturally occurring in the hexagonal unit cell. | ||||||
| 2H (wurtzite) | 1.24 | 3.21 | 11.78 | 4.29 | 6.28 | 15.10 |
| 4H | 1.20 | 3.15 | 11.67 | 2.37 | 4.25 | 12.59 |
| 5H | 1.16 | 3.12 | 11.68 | 1.91 | 3.79 | 12.20 |
| 6H | 1.17 | 3.11 | 11.58 | 1.79 | 3.66 | 12.11 |
| 8H | 1.14 | 3.09 | 11.58 | 1.54 | 3.44 | 11.87 |
| 9Ra | 1.19 | 3.16 | 11.66 | 2.13 | 4.04 | 12.48 |
| 12Ra | 1.10 | 3.08 | 11.59 | 1.71 | 3.60 | 12.00 |
| 15Ra | 1.14 | 3.13 | 11.57 | 1.59 | 3.48 | 11.91 |
| 3C (sphalerite) | 1.12 | 3.08 | 11.57 | — | — | — |
We can confirm the previous experimental and theoretical investigations of zinc oxide, where the primary direct band gap is observed at the Γ-point of the Brillouin zone. Sphalerite (3C), has the smallest gap of all the ZnO polytpes and wurtzite (2H) the largest one, computed with B3LYP and Hartree–Fock. Furthermore, an increase of the fraction of hcp-layers in the hexagonal polytypes (also known as hexagonality in the literature52) usually results in a larger band gap.
In addition, a secondary band gap was observed for the first time in zinc oxide, and a direct correlation between the stacking order and the secondary band gaps is shown in Table 2. We observe similar trends of widening the secondary gap, as for the primary band gap; with the increase of hexagonality the band gap widens. This increase of the band gap is especially noticeable between the 2H and the 4H polytype, independently of the calculational method employed (see Table 2).
4. Discussion
In Fig. 3, we show the band structures of the 4H and 15R polytype calculated using the B3LYP functional. Both polytypes show a primary band gap at the Γ-point; however, the 4H polytype shows the secondary gap at the A point, and the 15R polytype along the H–K direction of the Brillouin zone. The complete set of band structures for ZnO polytypes calculated with various ab initio methods are given in the ESI.†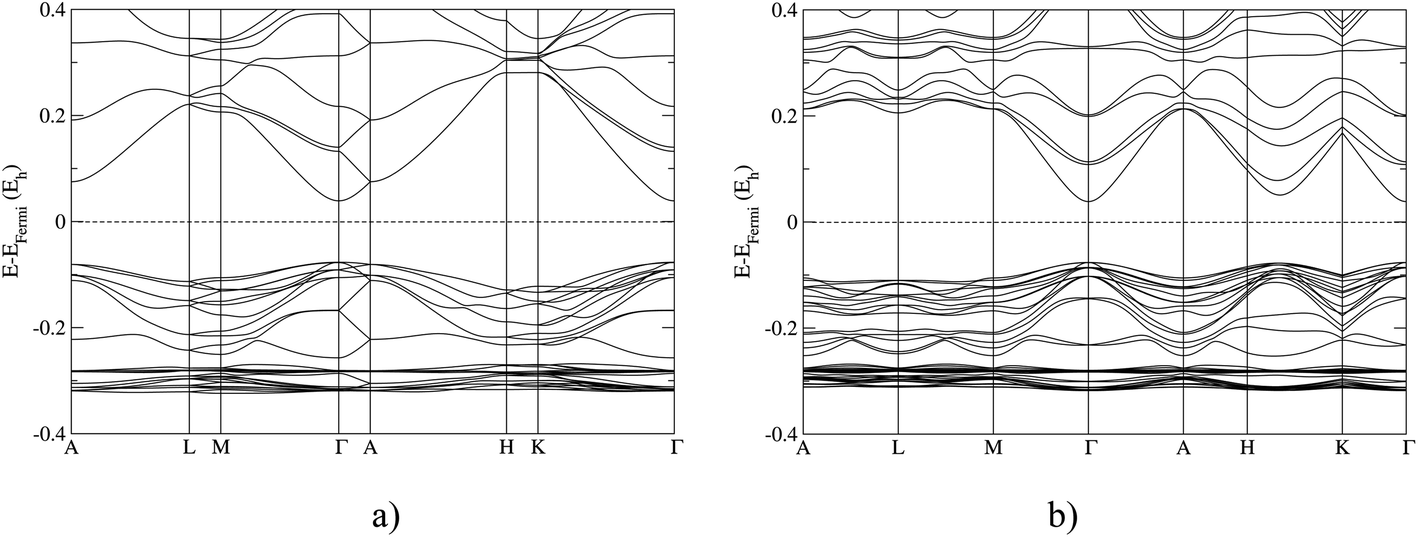 | ||
| Fig. 3 Band structure calculation using the hybrid B3LYP functional for (a) the 4H polytype; (b) the 15R polytype. Note that the labels of the special points of the Brillouin zones correspond to those of a hexagonal lattice. The rhombohedral unit cell is converted to a hexagonal one for comparison. | ||
There have been many examples in various systems where different stacking orders, based on mixing the hcp- and the fcc-type stacking, produce different polytypes.1–6 Our theoretical landscape explorations have also resulted in several interesting ZnO candidates which exhibit different stacking orders similar to the many polytypes in the ZnS system, while varying the numbers of formula units per simulation cell. Structurally, we can relate our polytype structures to those of zinc sulfide, or to the structures in silicon carbide, where different polytypes have also been observed.2–4,42,43,53,54
Concerning the effects of polytypism on the electronic structure, it is known that e.g. in silicon carbide (SiC) the 3C polytype shows the highest electron mobility.4,55 Furthermore, the band gaps of SiC differ widely among the polytypes ranging from 2.3 eV for the cubic 3C polytype (i.e. sphalerite) to 3.3 eV for the hexagonal 2H polytype (i.e. wurtzite), where the experimental polytpyes investigated have band gaps in-between (e.g. the 6H polytype has a gap of 3 eV). But quite generally, one finds that the larger the fraction of wurtzite-like components (2H), the larger the band gap.4,55 Our results show the same overall trends for the zinc oxide polytypes as in silicon carbide: sphalerite (3C) has the smallest gap of all the ZnO polytpes and wurtzite (2H) the largest, respectively, and by increasing the size of hcp layer regions one increases the band gap. Therefore, it should be possible, by combining fcc-type and hcp-type layer stackings, to reduce the size of the band gap (see Table 2). Note that in ZnO the 12R and 15R structures differ slightly from the 12R and 15R polytypes in ZnS, respectively, and this may also influence the band gap (for the structure data, cf. ESI†). Similar trends in the band gaps are observed in other III–V compounds.5
Clearly, the electronic effects of changing the stacking order in zinc oxide are not as dramatic as in silicon carbide. Nevertheless, this indicates that it is possible to tune the direct primary and secondary band gaps in ZnO to values between those of the wurtzite- and the sphalerite-modification by selecting the appropriate polytype without adding dopant atoms. Of course, the existence of polytypes makes it nontrivial to synthesize a macroscopic single-crystal; however if one can establish a degree of control during crystal growth sufficient to synthesize specific different polytypes, new materials for electronic device applications would be accessible.4,5,55 Recent attempts to generate nanofilms with unusual stacking orders9 are quite encouraging in this respect.
We note that the structure chemistry of zinc oxide and zinc sulfide appear to be quite similar. This similarity is demonstrated not only by the polytype calculations presented here, but also by many experimental observations, such as the existence of wurtzite and sphalerite modifications in both ZnO and ZnS, and recent investigations that suggest the occurrence of calculated,56 synthetic57,58 and natural59 polymorph of ZnO intergrown with ZnS. Therefore, we would like to comment on an ongoing discussion regarding the structural classification of the zinc sulfide mineral matraite, which has been claimed to be a 3R-polytype of ZnS. The most common polytypes of zinc sulfide are 2H (wurtzite) and 3C (sphalerite), while others are rarely encountered in nature.2,54,60 The purported 3R polytype was for the first time experimentally synthesized in the U.S. almost sixty years ago,61 while its natural form as the mineral matraite has been found in Hungary,62 and later in Bulgaria.63 However, as alternatives, a 6H polytype60 or a twinned sphalerite structure have been suggested42,59,64 as possible models for the matraite structure. Now, if such a 3R-structure modification were to exist as a distinct polytype, we would expect to observe it also in ZnO during our nearly exhaustive threshold investigations. This not being the case, we have constructed the 3R-type according to the description in ref. 61–63, and compared this structure with the ones found during the global searches. This structure comparison using the CMPZ algorithm65 clearly shows that the 3R polytype is equal to the 3C polytype both in ZnO and in ZnS, which would agree with the experimental results in ref. 42, 59 and 64. In addition, the structural-crystallographic constraints in the ZnO/ZnS compounds (or any analogous AB compound) show that the distances between the layers are the same as in the fcc packing, and therefore 3C must be equal to 3R. An exception would only be possible, if some special feature of the electronic structure of zinc oxide or zinc sulfide were to favor a second (structurally anisotropic) energy minimum right next to the sphalerite structure minimum on the energy landscape. None of our ab initio calculations suggest the existence of such a double minimum basin. While recent work on elemental zinc66 has shown that zinc can exhibit several electronic minimum energy configurations which induce slightly different atom positions, the bonding situation in zinc oxide/sulfide is completely different, and the presence of electron correlations strong enough to generate a second minimum is very unlikely. This is the reason, why in our global searches we observe only the 3C and 9R polytypes but not a separate 3R polytype (see Fig. 1). We would thus conclude that the matraite mineral is not a separate modification, but rather a (possibly twinned) 3C (sphalerite) polytype of ZnS.
5. Conclusions
To summarize, in order to gain new insights into the polytypism of the ZnO compound, we have performed structure prediction using simulated annealing and threshold runs with an empirical potential, followed by local minimizations on ab initio level. We have found the experimentally known structure types (wurtzite, sphalerite and rock-salt), in agreement with previous research, and we observe new low-energy polytypes of zinc oxide, which should be accessible to the experiment. A second focus of this research has been the investigation of the electronic properties of the newly discovered polytypes. We observe the same trends in the values of the band gap as in silicon carbide and zinc sulfide, and we note that the secondary band gap plays a role in zinc oxide polytypes. Clearly, these results suggest important new avenues in the development and fine-tuning of electronic devices with possible industrial applications. Additionally, we have clarified the ongoing discussion regarding the matraite (3R) mineral in zinc sulfide, which we can describe as a (possibly twinned) sphalerite (3C) structure.Acknowledgements
The authors would like to thank A. Recnik, J. Nuss, K. Doll, B. Matovic, K. Djuris and U. Wedig for valuable discussions.References
- U. Müller, Inorganic Structural Chemistry, Wiley-VCH, Marburg, Germany, 2007 Search PubMed.
- S. Mardix, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8677 CrossRef CAS.
- J.-S. Lee, S.-C. Yu, S.-F. Tung, W.-J. Bai, J.-S. Yang, Q.-S. Fang and Z. Zhang, Z. Kristallogr., 2006, 221, 213 CAS.
- H. Morkoc, S. Strite, G. B. Gao, M. E. Lin, B. Sverdlov and M. Burns, J. Appl. Phys., 1994, 76, 1363 CrossRef CAS.
- F. Bechstedt and A. Belabbes, J. Phys.: Condens. Matter, 2011, 25, 273201 CrossRef PubMed.
- S. Nakashima, H. Katahama, Y. Nakakura, A. Mitsuishi and B. Palosz, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 6531 CrossRef CAS.
- Ü. Özgür, Ya. I. Alivov, C. Liu, A. Teke, M. A. Reshchikov, S. Doğan, V. Avrutin, S.-J. Cho and H. Morkoç, J. Appl. Phys., 2005, 98, 041301 CrossRef.
- D. Zagorac, J. C. Schön, I. Pentin and M. Jansen, Process. Appl. Ceram., 2011, 5, 73 CrossRef CAS.
- M. Liu, G.-B. Ma, X. Xiong, Z.-W. Wang, Ru-W. Peng, J.-G. Zheng, Da-J. Shu, Z. Zhang and M. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 085306 CrossRef.
- J. C. Schön and M. Jansen, Angew. Chem., Int. Ed. Engl., 1996, 35, 1268 CrossRef.
- S. M. Woodley and C. R. A. Catlow, Nat. Mater., 2008, 7, 937 CrossRef CAS PubMed.
- D. Zagorac, J. C. Schön, J. Zagorac and M. Jansen, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 075201 CrossRef.
- D. Zagorac, J. C. Schön, J. Zagorac, I. V. Pentin and M. Jansen, Process. Appl. Ceram., 2013, 7, 111 CrossRef CAS.
- J. C. Schön, H. Putz and M. Jansen, J. Phys.: Condens. Matter, 1996, 8, 143 CrossRef.
- J. C. Schön, K. Doll and M. Jansen, Phys. Status Solidi B, 2010, 247, 23 CrossRef.
- S. Kirkpatrick, C. D. Gelatt and M. P. Vecchi, Science, 1983, 220, 671 CAS.
- S. W. de Leeuw, J. W. Perram and E. R. Smith, Proc. R. Soc. London, Ser. A, 1980, 373, 27 CrossRef CAS.
- R. Dovesi, R. Orlando, B. Civalleri, C. Roetti, V. R. Saunders and C. M. Zicovich-Wilson, Z. Kristallogr., 2005, 220, 571 CAS.
- K. Doll, R. Dovesi and R. Orlando, Theor. Chem. Acc., 2004, 112, 394 CrossRef CAS.
- A. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- J. C. Schön, Z. P. Cancarevic and M. Jansen, J. Chem. Phys., 2004, 121, 2289 CrossRef PubMed.
- D. Zagorac, K. Doll, J. C. Schön and M. Jansen, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 045206 CrossRef.
- J. E. Jaffe and A. C. Hess, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 7903 CrossRef CAS.
- T. Homann, U. Hotje, M. Binnewies, A. Borger, K. D. Becker and T. Bredow, Solid State Sci., 2006, 8, 44 CrossRef CAS.
- M. D. Towler, N. L. Allan, N. M. Harrison, V. R. Saunders, W. C. Mackrodt and E. Apra, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 5041 CrossRef CAS.
- A. M. Ferrari and C. Pisani, J. Phys. Chem., 2006, 110, 7909 CrossRef CAS PubMed.
- R. Hundt, KPLOT program-Version 9.6.15, University of Bonn, Bonn, Germany, 2012 Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653 CrossRef CAS.
- J. C. Schön, Z. Anorg. Allg. Chem., 2004, 630, 2354 CrossRef.
- Z. Cancarevic, J. C. Schön and M. Jansen, Chem.–Asian J., 2008, 3, 561 CrossRef CAS PubMed.
- C. H. Bates, W. B. White and R. Roy, Science, 1962, 137, 993 CAS.
- H. Liu, Y. Ding, M. Somayazulu, J. Qian, J. Shu, D. Häuserman and H.-K. Mao, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 212103 CrossRef.
- F. Decremps, J. Zhang, B. Li and R. C. Liebermann, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 224105 CrossRef.
- S.-K. Kim, S.-Y. Jeong and C. R. Cho, Appl. Phys. Lett., 2003, 82, 562 CrossRef CAS.
- J. E. Jaffe, J. A. Snyder, Z. Lin and A. C. Hess, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 1660 CrossRef CAS.
- D. Zagorac, J. C. Schön and M. Jansen, J. Phys. Chem. C, 2012, 116, 16726 CAS.
- C. R. A. Catlow, S. A. French, A. A. Sokol, A. A. Al- Sunaidi and S. M. Woodley, J. Comput. Chem., 2008, 29, 2234 CrossRef CAS PubMed.
- A. Kawska, P. Duchstein, O. Hochrein and D. Zahn, Nano Lett., 2008, 8, 2336 CrossRef CAS PubMed.
- M. Hellström, K. Jorner, M. Bryngelsson, S. E. Huber, J. Kullgren, T. Frauenheim and P. Broqvist, J. Phys. Chem. C, 2013, 117, 17004 Search PubMed.
- M. R. Farrow, Y. Chow and S. M. Woodley, Phys. Chem. Chem. Phys., 2014, 16, 21119 RSC.
- B. Meyer and D. Marx, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 035403 CrossRef.
- S. Haussuehl and G. Müller, Beitr. Mineral. Petrogr., 1963, 9, 28 CAS.
- R. G. Gevorkyan, G. A. Gurkina and F. V. Kaminskii, Am. Mineral., 1976, 61, 1054 Search PubMed.
- D. Milivojevic, B. Babic-Stojic and J. Kovac, Appl. Surf. Sci., 2010, 257, 937 CrossRef CAS.
- K. Siraj, K. Javaid, J. D. Pedarnig, M. A. Bodea and S. Naseem, J. Alloys Compd., 2013, 563, 280 CrossRef CAS.
- Z. Zeng, C. S. Garoufalis, S. Baskoutas and G. Bester, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 125302 CrossRef.
- J. Wrobel and J. Piechota, Solid State Commun., 2008, 146, 324 CrossRef CAS.
- Z. Li, Y. Xu, G. Gao, T. Cui and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 193201 CrossRef.
- S. H. Jang and S. F. Chichibu, J. Appl. Phys., 2012, 112, 073503 CrossRef.
- I. Demiroglu, S. Tosoni, F. Illasa and S. T. Bromley, Nanoscale, 2014, 6, 1181 RSC.
- L. Sponza, J. Goniakowski and C. Noguera, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 075126 CrossRef.
- K. Dornberger-Schiff and H. Schmittler, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1971, 27, 216 CrossRef CAS.
- H. Schulz and K. H. Thiemann, Solid State Commun., 1979, 32, 783 CrossRef CAS.
- G. Y. Chao and R. A. Gault, Can. Mineral., 1998, 36, 775 CAS.
- M. Bhatnagar and B. J. Baliga, IEEE Trans. Electron Devices, 1993, 40, 645 CrossRef CAS.
- J. Goclon and B. Meyer, Phys. Chem. Chem. Phys., 2013, 15, 8373 RSC.
- M.-Y. Lu, J. Song, M.-P. Lu, C.-Y. Lee, L.-Y. Chen and Z. L. Wang, ACS Nano, 2009, 3, 357 CrossRef CAS PubMed.
- G. Homm, et al. , J. Electron. Mater., 2010, 39, 1504 CrossRef CAS.
- V. Srot, A. Recnik, C. Scheu, S. Sturm and B. Mirtic, Am. Mineral., 2003, 88, 1809 CrossRef CAS.
- M. Pósfai and P. R. Buseck, Modular structures in sulphides: sphalerite/wurtzite-, pyrite/marcasite-, and pyrrhotite-type minerals, in EMU Notes in Mineralogy: Modular Aspects of Minerals, ed. S. Merlino, European Mineralogical Union (EMU), Budapest, Hungary, 1997, vol. 1, pp. 193–235 Search PubMed.
- D. C. Buck and L. W. Strock, Am. Mineral., 1955, 40, 192 Search PubMed.
- S. Koch, Acta Mineral.–Petrogr., 1958, 11, 11 CAS.
- J. A. Minceva-Stefanova, Mineral. Petrol., 1993, 49, 119 CrossRef CAS.
- E. Nitta, et al. , J. Mineral. Petrol. Sci., 2008, 103, 145 CrossRef CAS.
- R. Hundt, J. C. Schön and M. Jansen, J. Appl. Crystallogr., 2006, 39, 6 CrossRef CAS.
- N. Gaston, D. Andrae, U. Wedig and M. Jansen, Phys. Rev. Lett., 2008, 100, 226404 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra16574h |
| This journal is © The Royal Society of Chemistry 2015 |