
Synthesis, characterization and visible light photocatalytic activity of Cr3+, Ce3+ and N co-doped TiO2 for the degradation of humic acid
S. G. Rashida,
M. A. Gondal*a,
A. Hameedbc,
M. Aslamb,
M. A. Dastageera,
Z. H. Yamania and
D. H. Anjumd
aLaser Research Group, Physics Department and Center of Excellence in Nanotechnology (CENT), King Fahd University of Petroleum & Minerals, Dhahran 31261, Saudi Arabia. E-mail: magondal@kfupm.edu.sa; Fax: +96638602293; Tel: +96638602351
bCenter of Excellence in Environmental Studies, King Abdulaziz University, Jeddah 21589, Saudi Arabia
cNational Centre for Physics, Quaid-e-Azam University, Islamabad 44000, Pakistan
dApplied Surface Science Nanofabrication, Imaging & Characterization Core Lab, King Abdullah University of Science & Technology (KAUST), Thuwal 23599-6900, Saudi Arabia
First published on 31st March 2015
Abstract
The synthesis, characterization and photocatalytic activity of Cr3+ and Ce3+ co-doped TiON (N-doped TiO2) for the degradation of humic acid with exposure to visible light is reported. The synthesized bimetal (Cr3+ + Ce3+) modified TiON (Cr–Ce/TiON), with an evaluated bandgap of 2.1 eV, exhibited an enhanced spectral response in the visible region as compared to pure and Ce3+ doped TiON (Ce/TiON). The XRD analysis revealed the insertion of Cr3+ and Ce3+ in the crystal lattice along with Ti4+ and N that resulted in the formation of a strained TiON anatase structure with an average crystallite size of ∼10 nm. Raman analysis also supported the formation of stressed rigid structures after bimetal doping. HRTEM confirmed the homogeneous distribution of both the doped metallic components in the crystal lattice of TiON without the formation of surface oxides of either Cr3+ or Ce3+. Electron energy loss spectroscopy (EELS) analysis revealed no change in the oxidation of either Cr or Ce during the synthesis. The synthesized Cr–Ce/TiON catalyst exhibited appreciable photocatalytic activity for the degradation of humic acid on exposure to visible light. Additionally, a noticeable mineralization of carbon rich humic acid was also witnessed. The photocatalytic activity of the synthesized catalyst was compared with pristine and Ce3+ doped TiON.
Introduction
The exceptional photocatalytic activity, chemical stability, non-toxicity, solar energy conversion, hydrogen production and numerous photocatalytic applications for environmental remediation have brought TiO2 into the limelight of material research. Despite all the attractive features and the capability to generate highly oxidizing reactive oxygen species (ROS), its inherent limitations such as wide band gap energy (3.2 eV) and high rate of electron–hole recombination, confines its use as an effective photocatalyst for widespread commercial applications. Ever since Fujishima et al., reported their pioneering work in 1972, many alterations have been attempted to overcome the above-mentioned limitations of titania to make it responsive in the visible region of the light spectrum without losing its intrinsic activity, and many non-metallic dopants have been tested, but none of these exhibited a substantial red shift in the band gap energy.1,2 Among the non-metal modified titania, N-doped TiO2 (TiON) is the only reported material with comparable activity in the visible region.3 The effectiveness of nitrogen as a dopant in TiO2 was attributed to the comparative size and electronegativity of nitrogen and oxygen atoms which resulted in the narrowing of the bandgap. Although N-doping of Titania shifts the bandgap in the visible region, however, doping with an anionic constituent results in low quantum efficiency and long-term instability.4,5Recently, among the metallic dopants, Ce3+ and Ce4+ ions, have been reported as effective dopants with the trapping and transfer ability and improving the quantum efficiency of TiO2.6–12 Considering these features of Ce3+ and the reduced bandgap energy of TiON, the modification of the TiON with Ce3+ may result in the enhanced activity and stability, however, no substantial red shift with substantial activity for visible light applications is documented. It has also been reported that Cr3+, as a modifier, can substantially improve the absorption of light in the visible region, however, for large band gap TiO2, chromium as a sole dopant exhibited detrimental effects,13,14 whereas, chromium as a co-dopants with nitrogen in titania exhibited an enhanced photocatalytic activity.15
Humic acids are complex fused ring organic macromolecules attached with a variety of functional groups such as amines, amides, carboxylic, carbonyl, hydroxyl and others. The presence of humic acids during the water treatment results in the formation of mutagenic trihalomethane that cause membrane fouling and plugging, boost the re-growth of bacteria in distribution systems and corrosion of pipelines.16–21 Based on the associated environmental issues, humic acid has been the substrate of interest in adsorption,22 photo-fenton,23 photo-electrocatalytic24 and photocatalytic25–27 studies. The comparison of the photocatalytic activity of TiO2 and non-TiO2 based photocatalysts for the degradation of humic acid under various experimental conditions is compared in Table 1.26,28–38
| S. no. | Photocatalyst | % Degradation/mineralization | Source | References |
|---|---|---|---|---|
| 1 | TiO2 | 88% | Simulated solar radiation | 28 |
| 2 | TiO2 | 90% | UV | 29 |
| 3 | TiO2 | 95% | UV | 30 |
| 4 | TiO2 | 65% | UV | 31 |
| 5 | TiO2 | 80% | UV | 32 |
| 6 | TiO2 | 52.5–83.1% | UV | 33 |
| 7 | TiO2 | 60% | UV | 34 |
| 8 | TiO2 | 52.5% | Solar simulator | 35 |
| 9 | Pt–TiO2 | 29.1% | Solar simulator | 35 |
| 10 | W–TiO2 | 11.7% | Solar simulator | 35 |
| 11 | Co–TiO2 | 8.9% | Solar simulator | 35 |
| 12 | CuXO/H2–TiO2 | 5.8% | Solar simulator | 35 |
| 13 | Fe–TiO2 | 5.5% | Solar simulator | 35 |
| 14 | Fe–TiO2/SAC | 32.54% | UV | 36 |
| 15 | Fe2O3/TiO2 | 61.58% | UV | 37 |
| 16 | TiO2/GAC | 99.5% | UV | 38 |
| 17 | Ag/ZnO | 100% | Natural sunlight | 26 |
Motivated by the above mentioned positive attributes of Ce–N co-doped titania and Cr–N co-doped titania, in the current study, we synthesized a multi component Cr–Ce/TiON nano-structured photocatalyst by a hybrid sol gel-hydrothermal technique. The optical, structural and morphological characterization of the synthesized catalyst was performed. Based on the environmental concerns associated, the photocatalytic efficiency of the newly synthesized Cr–Ce/TiON was assessed for the degradation of humic acid in the visible region (420–800 nm) using visible band pass filter and compared with TiO2 (P-25), TiON and Ce/TiON. The existence of absorption bands of humic acid at 277 nm and 370 nm was advantageous as it ruled out the possibility of sensitization by the humic acid molecules and the actual activity of the catalyst in the visible light exposure might be observed.
Experimental details
Synthesis of photocatalyst
Cr–Ce/TiON nanoparticles were synthesized by adopting the hybrid sol gel-hydrothermal technique.39,40 The chemicals used for synthesis of Cr–Ce/TiON were titanium isopropoxide (TIPO) (Sigma-Aldrich, 97%), urea (Sigma-Aldrich, 99.9%), cerium(III) nitrate hexahydrate (Sigma-Aldrich, 99.9%), n-hexane (Sigma-Aldrich, 99.9%), chromium(III) nitrate nonahydrate (Sigma-Aldrich, 99.9%), anhydrous ethanol (Merck, 99.9%), acetic acid (Sigma-Aldrich, 99.7%). In the typical synthesis of Cr–Ce/TiON, 0.2 g of cerium(III) nitrate hexahydrate and 0.6 g chromium(III) nitrate nonahydrate were dissolved in a mixture of solvents of ethanol, acetic acid and water in 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 ratio at 10 °C. The appropriate amount of urea was added as a nitrogen source. Under vigorous stirring (1500–2000 rpm), 18.5 ml of TIPO was added drop-wise and left for 12 h in the dark for nucleation. The suspension was placed in an oven at 200 °C temperature for 6 h in a Teflon® coated autoclave. The resultant slurry was filtered, washed with ethanol, dried at room temperature and calcined at 400 °C for 3 h in a muffle furnace at a heating rate of 1°C min−1. The powder was allowed to cool at the cooling rate of 1 °C min−1 till room temperature. The similar methodology was adopted for the synthesis of TiON and Ce/TiON nanoparticles.
:2:1 ratio at 10 °C. The appropriate amount of urea was added as a nitrogen source. Under vigorous stirring (1500–2000 rpm), 18.5 ml of TIPO was added drop-wise and left for 12 h in the dark for nucleation. The suspension was placed in an oven at 200 °C temperature for 6 h in a Teflon® coated autoclave. The resultant slurry was filtered, washed with ethanol, dried at room temperature and calcined at 400 °C for 3 h in a muffle furnace at a heating rate of 1°C min−1. The powder was allowed to cool at the cooling rate of 1 °C min−1 till room temperature. The similar methodology was adopted for the synthesis of TiON and Ce/TiON nanoparticles.
Characterizations of photocatalyst
The structural characterization of the synthesized nanoparticles was performed with wide angle X-ray diffractometer (Philips X'Pert PRO 3040/60) equipped with Cu-Kα radiation source in the 2θ range of 10 to 80 degrees. The transmission electron microscopy (TEM) images were recorded with Titan G2 80-300 (FEI Company, Hillsboro, USA), operated at a primary beam energy of 300 keV, while point-to-point analysis was performed at the same beam energy with a step of 0.235 nm. A charge-coupled device (CCD) camera (US4000, Gatan, Inc., Pleasanton, CA) was used to record digital images. Furthermore, the crystal structure and the morphological phases of the nanoparticles in Cr–Ce/TiON samples were extracted by high resolution TEM (HRTEM) analysis for the confirmation of XRD results. The elemental composition and oxidation state analysis of the components of the bimetal doped TiON was performed by using energy filter Tridiem™ 683 (Gatan, Inc., Pleasanton, CA). The electron energy loss spectra (EELS) were also recorded during the course of TEM analysis. Entire image processing of HRTEM and STEM-EELS SI datasets were carried out in Gatan Microscopy Suite (GMS) version 1.8.3. The optical bandgaps of the synthesized powders were evaluated by applying Kubelka–Munk transformation on the reflectance data recorded by Jasco 570 UV-Vis diffuse reflectance spectrophotometer. The Brunauer–Emmett–Teller (BET) surface areas of the synthesized powders were measured by N2-adsorption at 77.4 K using Quntachrome NovaWin 2. Prior to adsorption measurements, the samples were degassed for 3 h at the temperature of 300 °C. The surface functional groups were identified by Spectrum-1000 of Perkin-Elmer FT-IR spectrometer. The Raman shifts were measured using a DXR Raman Microscope, Thermo Scientific, USA, equipped with 532 nm laser as the excitation source at 6 mW power.The photocatalytic activity of Cr–Ce/TiON, in comparison to pristine and Ce/TiON, was evaluated for the degradation of humic acid in a visible light exposure. A 1000 ppm stock solution of humic acid was prepared by dissolving 1000 mg of humic acid initially in 20 ml of 1 M NaOH solution and finally diluted to 1000 ml. Photocatalytic experiments were performed in batches. In a typical experiment, 100 ml of catalyst/humic acid suspension containing 50 ppm humic acid and 100 mg of the synthesized catalysts, was exposed to visible light (420–800 nm) in a cylindrical Pyrex glass reactor equipped with stirrer. The visible light was generated by the 450 W xenon arc lamp equipped with visible light band pass filter. Prior to exposure, the suspension was stirred in the dark for 30 min for establishing of adsorption–desorption equilibrium. The samples were drawn at the regular interval of 30 min in the first 2 h and after 60 min in the final hours. After removing the catalyst by 0.22 μm Whatman syringe filter, the samples were analysed by UV-visible spectrophotometer and TOC (Shimadzu Corporation, Japan) for the residual humic acid concentration and carbon measurements, respectively. The fluorescence of the samples was measured by Horiba Fluorolog®-3 Spectroflurometer. The degradation of humic acid was calculated at 277 and 370 nm, absorption maxima of humic acid, as a function of time. Blank experiments were also performed to evaluate the possible effects of interaction of visible photons with humic acid.
Results and discussion
The comparison of the XRD profiles of the synthesized TiON, Ce/TiON and Cr–Ce/TiON powders are presented in Fig. 1. The reflections due to Ce/TiON and Cr–Ce/TiON were matched with that of pure TiON. Except for the reflection of (004) face, all the other reflections were matched with pure TiON and appeared at 2θ values of 25.62, 48.40, 54.54, 63.28, 67.68 and 74.71 degrees. As presented in the inset of Fig. 1, the reflections of (004) face for TiON, Ce/TiON and Cr–Ce/TiON appeared at 38.04, 38.14 and 38.44 degrees, respectively. The variation in the peak position may be attributed to the ionic radii of the dopants involved and an indication of the formation of the stressed lattice. The hkl indices corresponding to the above mentioned reflections are presented on each reflection in Fig. 1. All the major and minor reflections of the synthesized powders were typical of TiO2 (anatase, JCPDS # 84-1286) indicating the phase purity of the synthesized powders. Additionally, the absence of any additional reflection corresponding the formation of surface off shoots of cerium or chromium, such as oxides also depicted the homogeneous distribution of the dopants in the lattice of TiON. The average crystalline sizes of Ce/TiON and Cr–Ce/TiON, evaluated by applying Scherer's relation on the most intense reflections were 10.1 nm and 9.2 nm, respectively.41 | ||
| Fig. 1 The comparison of the XRD patterns of Ce/TiON (black) and Cr–Cr/TiON (black) in the range of 2θ = 10° to 2θ = 80°. The hkl indices of the corresponding reflection are mentioned on each reflection. The inset shows the variation in the position of (004) reflection with the insertion of Ce3+ and Cr3+ + Ce3+ in TiON matrix. | ||
The HRTEM electron micrograph of the synthesized powder is presented in Fig. 2(A) where the particles of almost uniform morphology are observable. The average particle size of the synthesized Cr–Ce/TiON nanoparticle was determined by measuring the size of about 200 particles imaged with HRTEM. The measured average particle size of ∼10 nm authenticated the average crystallite size calculated from XRD measurements and confirmed the existence of Cr–Ce/TiON nanoparticles as nano-range single crystals. As presented in Fig. 2(B), the crystal structure determination of Cr–Ce/TiON nanoparticles was performed by applying the Fast Fourier Transformation (FFT) of the HRTEM electron micrograph presented in Fig. 2(A). The concentric rings on the FFT of micrograph image represent the diffraction peaks observed in the XRD pattern (Fig. 1). The radii of the rings were directly related to dhkl-spacing measured by applying Circular Hough Transformation (CHT) to the FFT data.42 As expected, these values of radii obtained from the analysis of FFTs are in agreement with the diffraction pattern of Cr–Ce/TiON measured from the X-ray diffraction technique and hence these rings can be indexed in the same way as the XRD peaks were indexed in Fig. 1. It was noticed that dhkl spacing determined by both XRD and HRTEM techniques was in good agreement with each other and confirmed the existence of Cr–Ce/TiON in anatase crystal phase. The EF-TEM mappings of the individual components of chromium (Cr) and cerium (Ce) doped TiON are presented in Fig. 3, where the homogeneous distribution of all dopants throughout titania matrix was observable without any unwanted irregular concentration of dopants.
 | ||
| Fig. 2 (A) The HRTEM micrograph (B) the Fast Fourier Transformation (FFT) of HRTEM micrograph of Cr and Ce doped TiON elaborating the respective hkl indices in terms of concentric circles. | ||
 | ||
| Fig. 3 The EF-TEM images showing the mapping/distribution of the Ti, Ce, Cr, O and N in Cr and Ce doped TiON. | ||
The TEM-EELS analysis of Cr–Ce/TiON was performed for the determination of elemental composition and the valence states of individual components. The EELS spectrum, acquired by setting the microscope in STEM mode, is presented in Fig. 4, where the presence of the edges due to the individual components confirms the adequate insertion of Cr3+, Ce3+ and N in TiO2 matrix. The quantified atomic percentages of Ti, O, N, Cr, and Ce in Cr–Ce/TiON were ∼37.74%, ∼58.89%, ∼1.36%, ∼0.95% and ∼1.06%, respectively. The EELS edges furnished by the individual component in Cr–Ce/TiON were identified and marked by dotted lines in Fig. 4. The edges due to Ti and Cr were of L23 while for Ce of M45 type. The L3 edges are originated as a result of the electron transitions from the inner 2p3/2 orbitals to empty 3d orbital, whereas the L2 edges originate due to the electronic transition from 2p1/2 to 3d orbitals. These L23 edges of 3d transition metals and M45 for 4d transition metals are termed as “white lines”. Being a function of LS/JJ coupling, the intensity of white-lines directly corresponds to the oxidation states of the transition metals involved. Therefore, to determine the valence states of the metals involved in the synthesis process, the intensities of the corresponding white-lines (Fig. 4) were extracted, normalized using the double-arc method43 and subsequently compared with the literature values. The adopted procedure revealed +3.7, +3 and +3.3 oxidation states for Ti, Cr and Ce respectively in Cr–Ce/TiON matrix. The oxidation state of 3.7 for Ti depicted the presence of a fraction in +3 oxidation state in anatase phase. This effect might be due to the combinatorial effect of N and Cr as both the dopants not only impart a negative charge to the system, but, being slightly larger in size than Ti4+ and O2−, generate size oriented strain that results in the loss of oxygen from the matrix. The oxygen vacancies generated contribute in the enhancement of spectral response and extended lifetime of the excited states. The variation in the oxidation state of Ce and the retention of oxidation states by Cr is beneficial in terms of photocatalytic activity.44–46
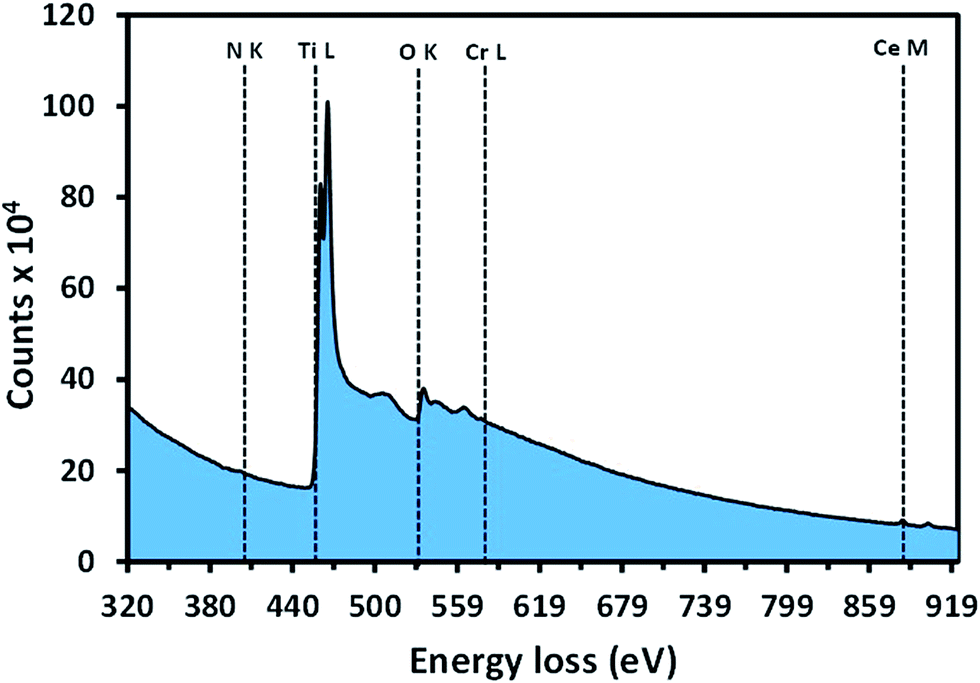 | ||
| Fig. 4 The EELS spectra of Cr and Ce doped TiON showing the edges yielded by the electronic transitions in N, Ti, O, Cr and Ce. | ||
The band gap energies of the synthesized materials (TiON, Ce/TiON and Cr–Ce/TiON) were estimated by applying Kubelka–Munk transformation on the reflectance data acquired by diffuse reflectance spectroscopy. The plot of [F(R) × hν]1/2 versus hν (photon energy) for the graphical determination of the bandgaps of the synthesized powders is presented in Fig. 5, where the band edge energies were evaluated by the extrapolation of the linear region of the plot starting from the absorption edge.47 From Fig. 5, the estimated band gap energies for TiON, Ce/TiON, and Cr–Ce/TiON were 3 eV, 2.8 eV and 2.1 eV, respectively. The systematic red shift in the band gap energies, with each modification eventually makes Cr–Ce/TiON absorptive in the visible region of the spectrum. In the case of Ce/TiON nanoparticles, the red shift of the band gap energy (3 eV) compared to pure titania (3.2 eV) is purely due to nitrogen doping.3 The interaction between the O2− and N, being non-metal and electronegative, results in the repositioning of the valence band at a relatively higher energy than pure TiO2. The mutual interaction between Ti4+, Ce3+ in Ce/TiON further contribute in the rearrangement of the conduction band constituted by Ti4+ in TiON that results in the enhanced absorption in visible region. The enhanced absorption of Cr–Ce/TiON is mainly due to the insertion of additional low energy bands by Cr3+ states and the effect charge transfer from Cr3+ to Ti4+ i.e. 4A2g → 4T1g and 4A2g → 4T2g d–d transition of Cr3+.48,49 It is also proposed that the insertion of impurity leads to the induction of interfacial bands below the conduction band of TiO2. The charge transfer from impurity band to the conduction band of TiO2 results in the enhanced absorption under illumination.48 However, in the current study, the smoothness of the diffuse reflectance spectra, the absence of additional absorption edges and the appearance of the single bandgap energy for the synthesized materials negated the formation of interfacial impurity induced bands therefore, the formation of a single conduction band with the mutual interaction of the dopants with Ti4+ is most likely. The same can be represented pictorially as under in Scheme 1.
 | ||
| Fig. 5 The graphical evaluation of the bandgap energies of TiON, Ce/TiON and Cr–Ce/TiON obtained by plotting (F(R) × hν)1/2 versus the photon energy hν. | ||
 | ||
| Scheme 1 The plausible rearrangement of valence and conduction band in TiO2 with the insertion of N, (N + Ce3+) and (N + Cr3+ + Ce3+). | ||
The FTIR spectrum of Cr–Ce/TiON in comparison to TiON and Ce/TiON was acquired to rule out the presence of organic residues. As presented in Fig. 6(a), the broad absorbance in the 3100–3500 cm−1 region was assigned to the OH stretching vibrations, whereas the strong band cantered at 1630 cm−1 was assigned to the vibrations of the surface-adsorbed H2O and Ti–OH bonds of Cr–Ce/TiON.50 A broad band in the range of 400–900 cm−1 was assigned to Ti–O stretching and Ti–O–Ti bridging stretching modes.51
 | ||
| Fig. 6 (a) The comparison of the FTIR spectra of TiON, Ce/TiON and Cr–Ce/TiON (b) the comparison of the Raman spectra of TiO2, TiON, Ce/TiON and Cr–Ce/TiON. | ||
The comparison of the Raman spectra of TiO2, TiON, Ce/TiON and Cr–Ce/TiON is presented in Fig. 6(b). In the current study, the Raman active modes in pure TiO2 (anatase) corresponding to B1g, A1g + B1g and Eg modes were appeared at 386.84 cm−1, 502.46 cm−1 and 628.75 cm−1, respectively. The observed values were in good agreement with the literature values.52 For TiON, the insertion of N in the lattice of TiO2 resulted in the expected shifting in B1g band to 380.44 cm−1 depicted that the presence of N in the lattice resulted in the increase in the rigidity of the system. The significant decrease in the intensity of the principal Raman bands mentioned above further elaborates the decreased magnitude of these vibrations with the insertion of Ce and Cr + Ce in Ce/TiON and Cr–Ce/TiON. This effect was again a credible verification of the presence of doped metals in the lattice of TiO2. Additionally the non-existence of additional bands verifies the phase purity without the formation of oxides of the respective metals.
BET surface areas of TiON, Ce/TiON and Cr–Ce/TiON nanoparticles calculated from the linear parts of the BET plots were 120 m2 g−1, 137 m2 g−1 and 141 m2 g−1, respectively. The observed enhancement in the surface area of the synthesized Ce/TiON and Cr–Ce/TiON compared to TiON make them more effective photo-catalysts in applications point of view.
Photocatalytic degradation of humic acid
In an aqueous phase photocatalytic system, the interaction of photons with the semiconductor particles results in the generation of variety of radical and ionic species, termed as reactive oxygen species (ROS) that either interact with the substrate or transform into molecular species by self-interaction. The primary oxidizing species, i.e. hydroxyl (HO˙) and super oxide anion radicals (O2˙−) are produced as a consequence of oxidation of the adsorbed H2O molecules and reduction of adsorbed or dissolved oxygen by the photon generated holes (h+) and electrons (e−), respectively. The process of formation of these radicals can be represented by the equations below.| H2O + hvb+ → HO˙ + H+ |
| O2 + ecb− → O2˙− |
It is believed that the interaction of these species does not lead to the mineralization of the organic substrates rather proceeds through the formation of intermediates.53–57 As illustrated below, the intermediates produced during the initial interaction of ROS with the substrates leading to the degradation, are further interacted by ROS for final conversion to CO2 and H2O.
| HO˙ + organic substrates → intermediates → CO2 + H2O |
| O2˙− + organic substrates → intermediates → CO2 + H2O |
We believe that the individual contribution of the ROS involved in the degradation and mineralization process may be assessed on the basis of the pace of degradation and mineralization (TOC removal) process.
Adsorption of the substrate at the surface of the photocatalysts is considered as an essential parameter that determines the interaction of photon generated ROS with the substrate molecules. In the current study, prior to the photocatalytic studies, the ability of the catalysts to adsorb the humic acid molecules, was evaluated. As presented in Fig. 7, it was noticed that the adsorption equilibrium was attained in ∼30 min of contact. The adsorption ability of both Ce/TiON and Cr–Ce/TiON was comparable, however, significantly higher than that of pure TiON. The higher adsorption of Ce and Cr + Ce modified TiON, as compared to pure TiON was attributed to the significant change in the surface area after the doping. The increasing surface area of the Ce (137 m2 g−1) and Cr + Ce (141 m2 g−1) doped TiON with increasing active sites, accommodate higher numbers of humic acid molecules compared to pure TiON (120 m2 g−1).
 | ||
| Fig. 7 The comparison of the time scale adsorption (mg g−1) of humic acid on TiON, Ce/TiON and Cr–Ce/TiON in dark. | ||
Based on the optical properties and that of humic acid, the photocatalytic performance of the synthesized Cr–Ce/TiON was evaluated in visible light exposure for the degradation/mineralization of humic acid. It is important to mention here that the blank experiments were performed to evaluate the degradation of humic acid by direct photolysis in visible light exposure however, no decrease in concentration of humic acid was noticed. The synthesized Cr–Ce/TiON catalysts possessed the bandgap of 2.09 eV (∼593 nm) whereas the substrate, humic acid, exhibited two strong absorption bands in the UV region centered at ∼277 nm and ∼370 nm with minimum absorption in the visible region that ruled out the possibility of photon absorption by the substrate, in competition to catalyst particles and lead to the evaluation of the photocatalytic activity exclusively by the catalyst particles. The decrease in the concentration of the humic acid was estimated by measuring the absorbance at 270 nm and 377 nm as this band measures the total aromaticity and the aromaticity due to the conjugation in condensed aromatic rings, respectively.58,59
The comparison of the time-scale percentage degradation profile for the degradation of humic acid (50 ppm) over P-25, TiON, Ce/TiON and Cr–Ce/TiON at 370 nm is presented in Fig. 8(a), where an enhanced activity of Cr–Ce/TiON compared to pure and Ce doped TiON is observable. Compared to ∼1.2, ∼8% and ∼23% for P-25, pure and Ce doped TiON, respectively, ∼52% decrease in the concentrations of humic acid was noticed in the initial 30 min of visible light exposure. With respect to the absorbance at 370 nm, the ∼99% removal was observed in 240 min of exposure, whereas the maximum removal of ∼11%, ∼43% and ∼79% was witnessed for P-25, pure and Ce doped TiON respectively, in the same period. The rate constants (min−1) for the degradation of humic acid over the three catalysts were evaluated by applying the Langmuir–Hinshelwood kinetic model for pseudo first order reactions. The graphical evaluation of the rate constants obtained by plotting ln(C0/C) versus the exposure time (t) is presented in Fig. 8(b). The evaluated rate constants for P-25, TiON, Ce–TiON and Cr–Ce/TiON were 0.0005 min−1, 0.0023 min−1, 0.0070 min−1 and 0.0187 min−1, respectively, with the average correlation factor of ∼0.97. The higher rate of degradation of humic acid over bimetal (Cr, Ce) doped TiON depicted the enhanced generation of ROS.
 | ||
| Fig. 8 (a) The comparison of the degradation of humic acid (50 ppm) over TiON, Ce/TiON and Cr–Ce/TiON in visible light exposure based on the decrease in the intensity of peak in UV-visible spectra at 370 nm (b) the graphical evaluation of the rate constants extracted by plotting ln(C0/C) versus visible light exposure time. | ||
The decrease in the total aromaticity of the humic acid was estimated by measuring the decrease in the intensity of the absorption band at 277 nm. The percentage degradation profile of humic acid over the catalysts under study, based on the decrease in the absorbance at 277 nm, is presented in Fig. 9(a), where a degradation pattern similar to that observed at 370 nm was noticed, however, the evaluated degradation with respect to the absorption at 370 nm, was significantly lower. For Cr–Ce/TiON, compared to ∼52% decrease in the concentration at 370 nm, ∼13% decrease in the concentration was witnessed in the initial 30 min of exposure whereas ∼91% removal was witnessed at the exposure time of 240 min. Similarly, significantly lower rates of degradation were noticed at 277 nm compared to 370 nm. As evaluated from Fig. 9(b), the values of rate constants for TiON, Ce–TiON and Cr–Ce/TiON were 0.0015 min−1, 0.0057 min−1 and 0.091 min−1, respectively, with the average correlation factor of ∼0.98. Although both A277 and A370 decreased, however, the relative intensities of the peaks at 277 nm and 370 nm (I277/I370) increased with the illumination time that furnished the elevated humic acid removal at 370 nm compared to 277 nm. In addition to absorption spectra, the removal of humic acid was also monitored by fluorescence spectroscopy. The representative fluorescence spectra showing the time scale decrease in the intensity with the decreasing concentration of humic acid over Cr–Ce/TiON is presented in the inset of Fig. 9(b). The peak center of the fluorescence spectrum is 436 nm and is attributed to polycyclic aromatics consisting of seven fused rings.60
 | ||
| Fig. 9 (a) The comparison of the degradation of humic acid (50 ppm) over TiON, Ce/TiON and Cr–Ce/TiON in visible light exposure based on the decrease in the intensity of peak in UV-visible spectra at 277 nm (b) the graphical evaluation of the rate constants extracted by plotting ln(C0/C) versus visible light exposure time. | ||
Owing to the higher bandgaps of 3.15 eV and 3.0 eV, the lower activity of P-25 and TiON for the degradation of humic acid may be attributed to its inability to absorb the visible photons which in turn affects the generation of ROS. Although the presence of Ce3+ in Ce/TiON significantly shifts the band gap in the visible region however a significant fraction of photons i.e. below 2.8 eV remains non-absorptive. For Cr–Ce/TiON, the presence of Cr3+ along with Ce3+ in the matrix of TiON, significantly lowers the bandgap (2.09 eV) and broadens its spectral response in the visible region compared to TiON and Ce/TiON. The lower bandgap energy, considerably increased the absorption of photons in the visible region that supports the significantly higher generation of reactive charge carriers for Cr–Ce/TiON. The superior degradation of humic acid demonstrates the ability of nanostructured Cr–Ce/TiON to transfer the charge carriers to adsorbed oxygen and H2O molecules for the generation of ROS. Here it can be speculated that the combined presence of Cr3+ and Ce3+ in the lattice of TiON induce the low energy states that facilitate the excitation of electrons in the visible light exposure initially and finally transferred to surface Ti4+ and Ti3+ states for onward transmission to reductants. With the equal probability of the presence of Cr3+ and Ce3+ at the surface, the direct transfer of excited electrons is equally probable.
The comparison of the TOC removal as a function of visible light illumination time during the degradation of humic acid over TiON, Ce/TiON and Cr–Ce/TiON nm is presented in Fig. 10(a). The pattern of the TOC removal was similar to that observed for the degradation, however, a significant decrease in the percentage TOC removal compared to degradation was noticed. In the initial 30 min of exposure, the TOC removal for TiON was negligible, whereas ∼7% and 12% TOC was removed for Ce/TiON and Cr–Ce/TiON that was significantly lower than the degradation of ∼23% and 51%, respectively. For Cr–Ce/TiON, compared to the degradation of ∼99% (370 nm) and ∼91% (277 nm) in 240 min of visible light exposure, ∼74% of TOC removal was witnessed. As presented in Fig. 10(b), a substantial decrease in the rate of TOC removal compared to degradation was noticed.
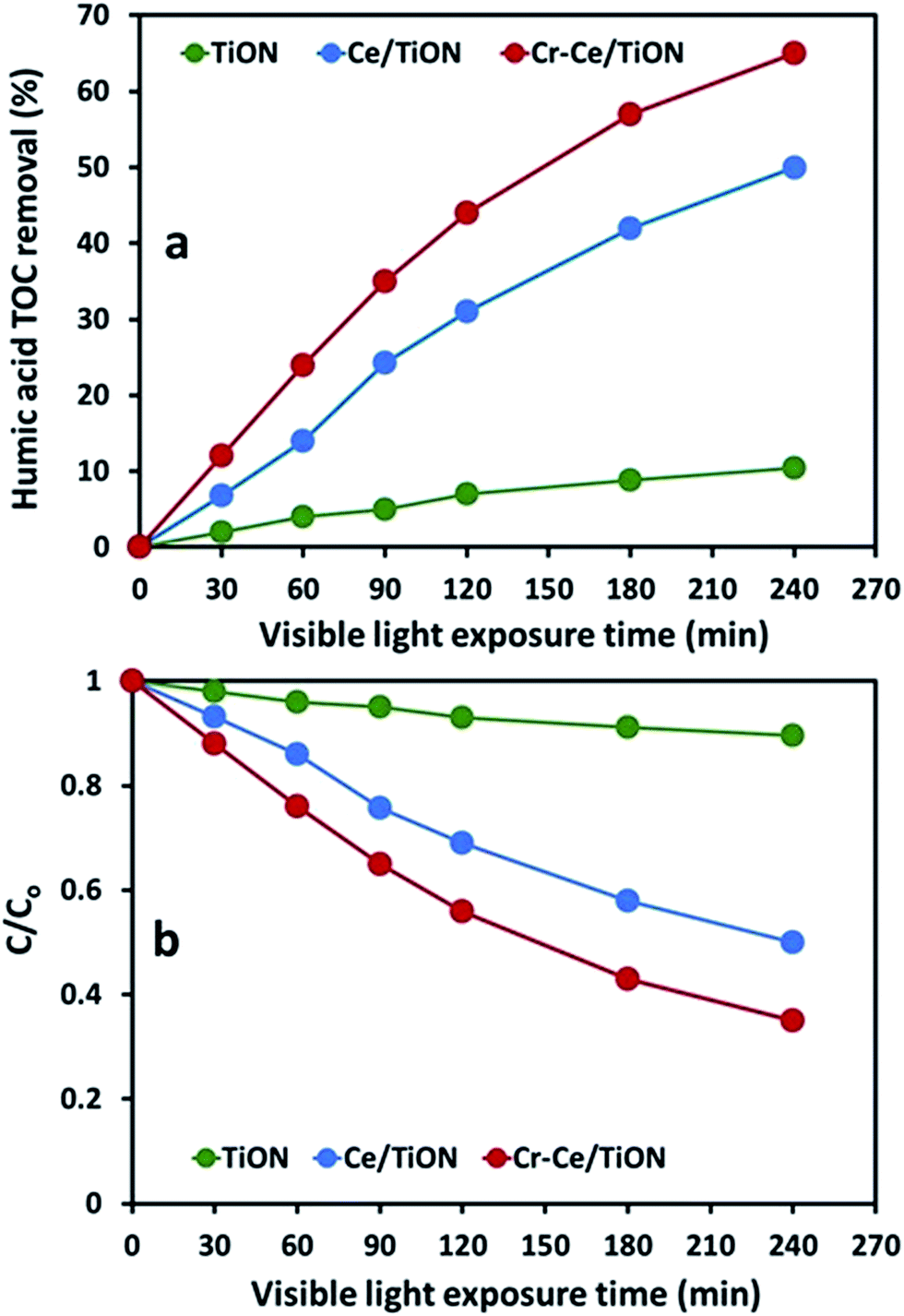 | ||
| Fig. 10 (a) The comparison of the TOC removal of humic acid (50 ppm) over TiON, Ce/TiON and Cr–Ce/TiON in visible light exposure (b) the C/C0 plot of the TOC removal versus visible light exposure time. | ||
The variation in the degradation rates at 277 nm and 370 nm lead to the conclusion that humic acid is degraded in a step wise mode. The initial interaction of ROS disturbs the conjugated structures that results in the formation of fragments with aromaticity that causes a significant decrease in the intensity of absorption band at 370 nm. However the formation of aromatic fragments restricts the proportional decrease in the intensity of the absorption band at 277 nm. Once the concentration of the humic acid is decreased substantially. The ROS interacts with the aromatic fragments formed initially that results in the decrease in the intensity of the band at 277 nm. The TOC removal further predicted that the larger components either aromatic or aliphatic are further subjected to oxidation till complete mineralization. The same can be represented by the Scheme 2 below.
 | ||
| Scheme 2 The plausible route of humic acid degradation and mineralization. | ||
The stability of the synthesized catalysts under visible light exposure was evaluated by comparing the XRD patterns of the exposed and unexposed catalysts. No significant change in the XRD patterns was noticed. A 3% acceptable variation in the activity of the catalysts was observed in three repeated exposures.
Conclusion
The study authenticated the suitability of the adopted procedure for the synthesis of doped multicomponent system. The study also revealed that the bimetallic doping of TiON not only increase the spectral response but also enhance the photocatalytic activity in the visible region of the spectrum however, the choice of the components is important. The enhanced photo-catalytic activity of Cr–Ce/TiON in the visible region may be attributed to the red shift in the band gap energy and +3 oxidation state of chromium. The doping by Ce and Cr not only increased the surface area of TiON but also enhanced the adsorption of substrates. The catalyst also showed the excellent ability to adsorb the substrate molecules. The rapid degradation of poly functional group containing carbon rich molecule revealed the generation significant number of ROS under visible light illumination over Cr–Ce/TiON. The interaction of the ROS produced under visible light illumination initially degrade the bulky humic acid molecules to smaller aromatic fragments that are further interacted by the ROS to complete mineralization. The synthesized catalyst may be regarded as addition to the existing photocatalysts.Acknowledgements
The support of the Department of Physics KFUPM and the Deanship of Scientific Research through MIT project # MIT11109 & MIT11110 is gratefully acknowledged. A. Hameed and M. Aslam acknowledge the support of Centre of Excellence in Environmental Studies (CEES), King Abdulaziz University and Ministry of Higher Education (MoHE), KSA.References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- S. G. Kumar and L. G. Devi, J. Phys. Chem. A, 2011, 115, 13211–13241 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- D. Li, H. Heneda, S. Hishata and N. Ohashi, Chem. Mater., 2005, 17, 2588–2595 CrossRef CAS.
- T. Yu, X. Tan, L. Zhao, Y. Yin, P. Chen and J. Wei, Chem. Eng. Technol., 2010, 157, 86–92 CrossRef CAS PubMed.
- X. Gao, X. S. Du, L. W. Cui, Y. C. Fu, Z. Y. Luo and K. F. Cen, Catal. Commun., 2010, 12, 255–258 CrossRef CAS PubMed.
- W. P. Shan, F. D. Liu, H. He, X. Y. Shi and C. B. Zhang, Appl. Catal., B, 2012, 115, 100–106 CrossRef PubMed.
- W. Cen, Y. Liu, Z. Wu, H. Wang and X. Weng, Phys. Chem. Chem. Phys., 2012, 14, 5769–5777 RSC.
- F. B. Li, X. Z. Li, M. F. Hou, K. W. Cheah and W. C. H. Choy, Appl. Catal., A, 2005, 285, 181–189 CrossRef CAS PubMed.
- J. M. Coronado, A. J. Maira, A. Martinez-Arias, J. C. Conesa and J. Soria, J. Photochem. Photobiol., A, 2002, 150, 213–221 CrossRef CAS.
- K. T. Ranjit, I. Willner, S. H. Bossmann and A. M. Braun, J. Catal., 2001, 204, 305–313 CrossRef CAS.
- T. Tong, J. Zhang, B. Tian, F. Chena, D. He and M. Anpo, J. Colloid Interface Sci., 2007, 315, 382–388 CrossRef CAS PubMed.
- D. Wang, J. Ye, T. Kako and T. Kimura, J. Phys. Chem. B, 2006, 110, 15824–15830 CrossRef CAS PubMed.
- J. Herrmann, New J. Chem., 2012, 36, 883–890 RSC.
- M. E. Kurtoglu, T. Longenbach, K. Sohlberg and Y. Gogotsi, J. Phys. Chem. C, 2011, 115, 17392–17399 CAS.
- M. L. Pacheco, E. M. Méndez and J. Havel, Chemosphere, 2003, 51, 95–108 CrossRef CAS.
- F. L. Palmer, B. R. Eggins and H. M. Coleman, J. Photochem. Photobiol., A, 2002, 148, 137–143 CrossRef CAS.
- M. Kulovaara, S. Metsamuuronen and M. Nystrom, Chemosphere, 1999, 38, 3485–3496 CrossRef CAS.
- A. K. Camper, Int. J. Food Microbiol., 2004, 92, 355–364 CrossRef CAS PubMed.
- X. Wang, Z. Wu, Y. Wang, W. Wang, X. Wang, Y. Bu and J. Zhao, J. Hazard. Mater., 2013, 262, 16–24 CrossRef CAS PubMed.
- R. Yuan, B. Zhoua, D. Hua and C. Shi, J. Hazard. Mater., 2013, 262, 161–167 CrossRef PubMed.
- K. Yang, D. Lin and B. Xing, Langmuir, 2009, 25, 3571–3576 CrossRef CAS PubMed.
- H. Katsumata, M. Sada, S. Kaneco, T. Suzuki, K. Ohta and Y. Yobiko, Chem. Eng. J., 2008, 137, 225–230 CrossRef CAS PubMed.
- X. Z. Li, F. B. Li, C. M. Fan and Y. P. Sun, Water Res., 2002, 36, 2215–2224 CrossRef CAS.
- A. Bansal, S. Madhavi, T. T. Y. Tan and T. M. Lim, Catal. Today, 2008, 131, 250–254 CrossRef CAS PubMed.
- M. Ghaneian, P. Morovati, M. Ehrampoush and M. Tabatabaee, J. Environ. Health Sci. Eng., 2014, 12, 138–144 CrossRef PubMed.
- X. Wang, Z. Wu, Y. Wang, W. Wang, X. Wang, Y. Bu and J. Zhao, J. Hazard. Mater., 2013, 262, 16–24 CrossRef CAS PubMed.
- J. Wiszniowski, D. Robert, J. Surmacz-Gorska, K. Miksch and J. V. Weber, J. Photochem. Photobiol., A, 2002, 152, 267–273 CrossRef CAS.
- M. Bekbolet, A. S. Suphandag and C. S. Uyguner, J. Photochem. Photobiol., A, 2002, 148, 121–128 CrossRef CAS.
- R. Al-Rasheed and D. J. Cardin, Chemosphere, 2003, 51, 925–933 CrossRef CAS.
- J. K. Yang and S. M. Lee, Chemosphere, 2006, 63, 1677–1684 CrossRef CAS PubMed.
- M. Lim, R. Fabris, C. Chow, K. Chiang, M. Drikas and R. Amal, Chemosphere, 2008, 72, 263–271 CrossRef PubMed.
- S. I. Patsios, V. C. Sarasidis and A. J. Karabelas, Sep. Purif. Technol., 2013, 104, 333–341 CrossRef CAS PubMed.
- Z. Yigit and H. Inan, Water, Air, Soil Pollut.: Focus, 2009, 9, 237–243 CrossRef CAS.
- D. Klauson, O. Budarnaja, I. C. Beltran, M. Krichevskaya and S. Preis, Environ. Technol., 2014, 35, 2237–2243 CrossRef CAS PubMed.
- M. H. Baek, J. S. Hong, J. W. Yoon and J. K. Suh, Int. J. Photoenergy, 2013, 2013, 5, DOI:10.1155/2013/296821.
- S. Qiao, D. D. Sun, J. H. Tay and C. Easton, Water Sci. Technol., 2003, 47, 211–217 CAS.
- G. Xue, H. Liu, Q. Chen, C. Hills, M. Tyrer and F. Innocent, J. Hazard. Mater., 2011, 186, 765–772 CrossRef CAS PubMed.
- Y. Cong, J. Zhang, F. Chen and M. Anpo, J. Phys. Chem. C, 2007, 111, 6976–6982 CAS.
- J. Zhang, Y. Wu, M. Xing, S. A. K. Leghari and S. Sajjad, Energy Environ. Sci., 2010, 25(3), 715–726 Search PubMed.
- C. S. Barret and T. B. Massalski, Structure of Metals, McGraw-Hill, New York, 1966 Search PubMed.
- D. R. G. Mitchell, Ultramicroscopy, 2008, 108, 367–374 CrossRef CAS PubMed.
- P. A. vanAken and B. Liebscher, Phys. Chem. Miner., 2002, 29, 188–200 CrossRef CAS PubMed.
- P. Reunchan, S. Ouyang, N. Umezawa, H. Xu, Y. Zhang and J. Ye, J. Mater. Chem. A, 2013, 1, 4221–4227 CAS.
- M. I. Ismail, M. Aslam, T. Almeelbi, S. Chandrasekaran and A. Hameed, RSC Adv., 2014, 4, 16043–16046 RSC.
- M. Aslam, I. M. I. Ismail, S. Chandrasekaran, T. Almeelbi and A. Hameed, RSC Adv., 2014, 4, 49347–49359 RSC.
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi, 1966, 15, 627–633 CrossRef CAS.
- Z. Liu, B. Guo, L. Hong and H. Jiang, J. Phys. Chem. Solids, 2005, 66, 161–167 CrossRef CAS PubMed.
- N. Serpone and D. Lawless, Langmuir, 1994, 10, 643–652 CrossRef CAS.
- Y. Liu, X. Wang, F. Yang and X. Yang, Microporous Mesoporous Mater., 2008, 114, 431–439 CrossRef CAS PubMed.
- M. M. Mohamed and K. S. Khairou, Microporous Mesoporous Mater., 2011, 142, 130–138 CrossRef CAS PubMed.
- H. C. Choi, Y. M. Jung and S. B. Kim, Bull. Korean Chem. Soc., 2004, 25, 426–428 CrossRef CAS.
- A. Hameed, M. Aslam, I. M. I. Ismail, S. Chandrasekran, M. Kadi and M. A. Gondal, Appl. Catal., B, 2014, 160–161, 227–239 CrossRef CAS PubMed.
- M. Aslam, I. M. I. Ismail, T. Almeelbi, S. Chandrasekaran and A. Hameed, Chemosphere, 2014, 117, 115–123 CrossRef CAS PubMed.
- M. Aslam, I. M. I. Ismail, S. Chandrasekaran and A. Hameed, J. Hazard. Mater., 2014, 276, 120–128 CrossRef CAS PubMed.
- A. Hameed, M. Aslam, I. M. I. Ismail, N. Salah and P. Fornasiero, Appl. Catal., B, 2015, 163, 444–451 CrossRef CAS PubMed.
- M. Aslam, I. M. I. Ismail, N. Salah, S. Chandrasekaran, M. T. Qamar and A. Hameed, J. Hazard. Mater., 2015, 286, 127–135 CrossRef CAS PubMed.
- S. J. Traina, J. Novak and N. E. Smeck, J. Environ. Qual., 1990, 19, 151–153 CrossRef CAS.
- K. H. Kang, H. S. Shin and H. Park, Water Res., 2002, 36, 4023–4032 CrossRef CAS.
- J. Peuravuori, R. Koivikko and K. Pihlaja, Water Res., 2002, 36, 4552–4562 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |