
Nb and Zr modified MWW zeolites – characterisation and catalytic activity
Anna Wojtaszek-Gurdak* and
Maria Ziolek
Adam Mickiewicz University in Poznań, Faculty of Chemistry, Umultowska 89b, 61-614 Poznań, Poland. E-mail: anna.gurdak@amu.edu.pl; Tel: +48 618291305
First published on 12th February 2015
Abstract
Hydrogen forms of MCM-22 and delaminated MCM-56 zeolites were modified with niobium and zirconium species. The structure/texture and surface properties, as well as the catalytic activity for dibutyl sulfide oxidation, of the two types of modified zeolites were compared. The presence of Brønsted (BAS) and Lewis (LAS) acid sites was detected after pyridine adsorption in all the prepared materials. The highest number of BAS and LAS was observed for HMCM-22. The number of BAS significantly decreased after metal modification because of the interaction between the metal sources and the BAS. External silanol groups of zeolites also participate in metal anchoring. The catalysts obtained were tested for liquid phase dibutyl sulfide oxidation with hydrogen peroxide. The texture of the zeolites and the nature of the metal modifier have a crucial effect on the oxidation of dibutyl sulfide. MCM-56 zeolites are more active than their counterparts of MCM-22 type. Application of niobium containing zeolites significantly increases the reaction rate, leading to 95% conversion after 3 h. The remarkable role of the Nb species is due to its interaction with H2O2 towards peroxo/superoxo species. Modification of both zeolites with zirconium does not increase their activity because of the strong interaction of zirconium species with sulfur compounds.
Introduction
Zeolites represent an important group of inorganic crystalline microporous materials used in numerous large-scale chemical technologies as heterogeneous catalysts thanks to their adjustable acidity, high-temperature stability, and shape-selectivity properties.1 Conventional zeolites are characterized by uninterrupted 3D framework structures. The new group of zeolites of MWW topology exhibits a layered 2D character. The most known zeolites in this group are MCM-22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2–6 and MCM-56.7 MCM-22 exhibits a structure similar to that found earlier for ERB-1,8 SSZ-259 or PSH-3.10 The layered zeolites are so-called two-dimensional (2D) zeolites. The characteristic feature of these zeolites is that the layers could be exfoliated before their fusion into a 3D framework. The 2D structure is associated with ordered zeolite frameworks formed through lamellar precursors.11 The preparation method can based on the swelling of MWW layered zeolite precursor followed by a number of different possible treatments. Depending on the way applied for transformation of swollen precursor one can obtain different zeolites like ITQ-212,13 and MCM-36.14 There are elegant review papers15–20 describing the history, methods of synthesis and properties of MWW zeolites. The interest in such zeolites is still growing.
2–6 and MCM-56.7 MCM-22 exhibits a structure similar to that found earlier for ERB-1,8 SSZ-259 or PSH-3.10 The layered zeolites are so-called two-dimensional (2D) zeolites. The characteristic feature of these zeolites is that the layers could be exfoliated before their fusion into a 3D framework. The 2D structure is associated with ordered zeolite frameworks formed through lamellar precursors.11 The preparation method can based on the swelling of MWW layered zeolite precursor followed by a number of different possible treatments. Depending on the way applied for transformation of swollen precursor one can obtain different zeolites like ITQ-212,13 and MCM-36.14 There are elegant review papers15–20 describing the history, methods of synthesis and properties of MWW zeolites. The interest in such zeolites is still growing.
MCM-22 and delaminated MCM-56 are members of the MWW zeolites family. The differences in the structure of these zeolites significantly affect their properties. Their unique crystal structure is formed by two independent 10-ring (10 MR) pore systems.3,21 The first one is sinusoidal and is localized inside the zeolite layers. The second one is formed upon fusion of the adjacent layers. Moreover, there are 12-ring cups or pockets on the surface layers.21,22 When the zeolite 3-D structure is formed, two pockets form a supercage (ca. 1.8 nm × 0.7 nm) inside the zeolite crystal. Such a space can be sufficient to host even bulky organic molecules. The disordering of zeolite layers, as in MCM-56 prepared by a one-pot synthesis procedure, can also enhance the accessibility to 12-ring pockets. The main difference between MCM-22 and MCM-56 zeolites is the connection of layers that make up their structure. MCM-22 is formed of layers that are stacked in an ordered manner while MCM-56 is made of very thin MWW monolayers and small crystals that are disordered.21 MCM-56 shares the same primary structure with MCM-22; however, it has a relatively low amount of supercages, but more 12-MR surface cups and acid sites exposed to the crystal exterior, and an easier accessibility to large organic molecules.23 Both zeolites MCM-22 and delaminated MCM-56 can be synthesized in a similar way, with the use of the same structure directing agent, which is hexamethyleneimine. The differences in synthesis are the Si/Al ratio and the crystallization time. MCM-56 contains more Al in comparison to MCM-22 zeolite. The final product can be obtained after two days of synthesis for MCM-56 (as opposed to ten days for MCM-22).21,24 Approximately 50% of BAS in MCM-56 can interact with a large base pivalonitrile compared to <30% for MCM-22.25 This behavior of MCM-56 seems to be very important for the catalytic transformation of bulky molecules.
MWW zeolites are important in material chemistry because of the possibility of diverse structural, textural and compositional modifications. The use of intercalated inorganic and/or organic guests allows the design and formation of new catalyst with unique properties.26 Modification with metals gives an opportunity to obtain bifunctional (acid-redox) catalysts. Wu et al. have for the first time synthesized the crystalline MWW precursor containing the individual zeolitic sheets simultaneously with both tetrahedrally coordinated boron and titanium.27 Ruan et al. speculated that Ti centres should occupy the pillars sites, which have a higher steric accessibility than network position in conventional 3D zeolites.28 Different layered precursors were prepared containing tetrahedrally coordinated metals incorporated into the framework of individual MWW layers, such as gallium,29 vanadium,30 and lanthanum.31 Recent reports in the literature indicate the new possibility of generating catalytic properties of MWW zeolites by stabilization as an interlayer expanded structure MWW-IEZ and modification with cerium. The obtained catalysts present high activity for the oxidation of CO to CO2 at room temperature.32
In this work, we have prepared hydrogen forms of MCM-22 and delaminated MCM-56 zeolites to be later modified with niobium or zirconium. The objective of our study was to comparatively examine MCM-22 and MCM-56 zeolites modified with Nb and Zr species. The structure/texture and surface properties as well as the catalytic activity for dibutyl sulfide oxidation with hydrogen peroxide were compared for the two types of zeolites modified with different metals (Nb and Zr). The choice of niobium and zirconium as modifiers of zeolites was dictated by the presence of both elements as neighbours in the same row of the periodic table but the different oxidation states of both metals are expected to generate different surface properties of the finally obtained catalysts based on both mentioned zeolites. One can expect different interactions of the Zr and Nb species with the reactants in the oxidation of dibutyl sulfide. At least one of the reactants has to be chemisorbed on the catalyst surface. Considering the reaction pathways and based on recent papers33,34 it can be expected favorable chemisorption of H2O2 on niobium species playing the role of active sites (the formation of peroxo species) or interaction with Brønsted acid sites towards the formation of active intermediates (hydrosuperoxo species). In turn, zirconium species are expected to interact preferentially with dibutyl sulfide.35 The use of these two different modifiers (Zr and Nb) allows us to draw a picture of the most favorable reaction pathway (via chemisorption of dibutyl sulfide or H2O2).
Zirconium containing zeolites are characterized by good stability and easy regeneration through calcination.36 They have widespread application in a wide range of reactions, especially in reactions requiring moderate acidity and oxidizing capabilities like hydrogenation, hydroxylation, oxidation or epoxidation.37 Niobium compounds exhibit special properties not shown by the compounds of neighbouring elements in the periodic table. Some of them, like stability or strong metal support interaction (SMSI) and strong metal oxide support interaction (SMOSI), are very important for a good quality catalyst.38–40 The characteristic features of niobium compounds are not only to act as sources of Lewis and Brønsted acidity but also the promoter effect and the support effect.38 Niobium oxides remarkably enhance catalytic activity and prolong catalyst life when they are added in small amounts to known catalysts. Niobium compounds have recently been widely studied as catalysts for various reactions. In the last few years they have been tested e.g. in the selective oxidation of hydrocarbons, oxidative dehydrogenation of propane, oxidation of alcohols and oxidation of organic sulfides.41 In the latter reaction, niobium species can play the role of an agent promoting the interaction of hydrogen peroxide with acidic hydroxyls towards active hydrosuperoxo species transformed to OH radicals.33 Therefore, MWW types of zeolites containing Brønsted acidity were chosen as supports for niobium and zirconium species.
In our previous paper42 we compared the textural and catalytic properties of MCM-22 and delaminated MCM-56 zeolites modified with molybdenum. This work is to some extent a continuation of the studies on MWW zeolites modified with other transition metals that are located in the same row of the periodic table (Nb and Zr). One can expect generation of different properties via modification by niobium and zirconium because of the various electronic states of both metals leading to various cationic charges and different interactions with hydrogen peroxide and sulfur containing compounds.
Experimental
Zeolite preparation
The MCM-22 and MCM-56 zeolites were synthesized using the same reagents: SiO2 (Ultrasil 3VN – Degussa), sodium aluminate (53% of Al2O3 and 42.5% of Na2O, Riedel-de Haen), distilled water, sodium hydroxide (Chempur), and hexamethyleneimine (HMI – Aldrich) as the template.The MCM-22 zeolite was prepared according to the procedure described in ref. 3. The synthesis gel had the following composition: SiO2/Al2O3 = 54, OH/SiO2 = 0.18, H2O/SiO2 = 44.9, Na/SiO2 = 0.18, HMI/SiO2 = 0.35. The synthesis mixture was stirred for 30 min at RT and then loaded into a Teflon-lined Parr reactor (300 cm3). Hydrothermal synthesis was carried out at 443 K for 40 h with continuous stirring. The product, MCM-22 precursor (MCM-22P), was recovered by filtration, washed with distilled water, and dried at 383 K.
The MCM-56 was synthesized according to the method proposed by Fung et al.7 with the synthesis gel composition: SiO2/Al2O3 = 23, OH/SiO2 = 0.21, H2O/SiO2 = 20, Na/SiO2 = 0.21, HMI/SiO2 = 0.35. The resulting synthesis gel was stirred for 30 min at RT. Hydrothermal synthesis was carried out in a Teflon-lined Parr reactor at 416 K for 48 h upon continuous stirring. The resulting catalyst (MCM-56) was filtered off, washed with distilled water and dried at 383 K.
To obtain the hydrogen forms of the MCM-22 and MCM-56 zeolites, the catalysts were modified by a twice-repeated ion exchange procedure at 323 K for 3 h using 0.5 M NH4NO3 solution. After ion exchange, the catalysts were dried at 373 K for 12 h and calcined at 823 K for 6 h.
Modification of zeolites with niobium and zirconium
Nb and Zr containing catalysts were prepared by an impregnation method using an aqueous solution of ammonium niobate(V) oxalate hydrate (Sigma-Aldrich) for Nb/HMCM-22 and Nb/HMCM-56 (5 wt% of Nb) and zirconium(IV) oxynitrate hydrate (Sigma-Aldrich) for Zr/HMCM-22 and Zr/HMCM-56 (5 wt% of Zr). HMCM-22 or HMCM-56 was placed in a flask connected to a rotary vacuum evaporator (BUCHI Rotavopar RII) and evaporated for 1 h. Next, a solution of niobium or zirconium source was added and the wet material was rotated (100 rpm) at 353 K for 10 h. At 353 K the evaporation started. After evaporation, the material was dried in an oven at 383 K for 8 h (heating rate 5 K min−1) and then calcined at 773 K for 4 h (heating rate 5 K min−1).Characterization methods
The materials prepared were characterized using XRD, N2 adsorption/desorption, UV-vis, and XPS.XRD measurements were carried out on a Bruker AXS D8 Advance diffractometer with Cu Kα radiation (λ = 0.154 nm) with a step size of 0.05° in the wide-angle range (6–60°).
N2 adsorption/desorption isotherms were obtained in a Quantachrome Instruments Autosorb iQ2 at 77 K. Samples were pretreated in situ under vacuum at 573 K. The surface area was calculated by the BET method. The volume of mesopores was determined from BJH, whereas the micropore surface area and micropore volume were determined by the t-plot method.
X-ray photoelectron spectroscopy (XPS) measurements were performed with a Specs photoelectron spectrometer equipped with a monochromatic microfocused Al Kα X-ray source (1486.7 eV).
UV-vis spectra were recorded on a Varian-Cary 300 Scan UV-visible spectrophotometer with an integrated sphere CA-30I. The catalysts, dehydrated at 673 K for 2 h, in the form of powders were placed into a cell equipped with a quartz window. The Kubelka–Munk function (F(R)) was used to convert the reflectance measurements into equivalent absorption spectra using the reflectance of SPECTRALON as a reference.
Acidity measurements
Infrared spectra were recorded with a Bruker Vector 22 FTIR spectrometer using an in situ cell. The samples were pressed under low pressure into thin wafers of ca. 10 mg cm−2 and placed inside the vacuum cell. Before pyridine adsorption, the catalysts were outgassed at 673 K for 2 h and pyridine was then admitted at 473 K. After saturation with pyridine, the samples were degassed at 473 K for 5 min. The catalysts were outgassed at different temperatures after pyridine adsorption. The spectrum without any sample (“background spectrum”) was subtracted from all recorded spectra. The spectra of the activated samples (after outgassing at 673 K) were subtracted from those recorded after the adsorption of pyridine followed by various treatments. The reported spectra are the results of this subtraction and are normalized to 10 mg cm−2.Cyclisation and dehydration of 2,5-hexanedione (2,5-HDN)
The catalysts were tested for 2,5-hexanedione (2,5-HDN) dehydration and cyclisation as the test reactions. A tubular, down-flow reactor (Ø = 8 mm; length l = 80 mm) was used for the 2,5-HDN dehydration and cyclisation reaction that was carried out at atmospheric pressure, using nitrogen as the carrier gas. The catalyst powder was pressed, crushed and sieved to the fraction of 0.5 < Ø < 1.0 mm. A total of 0.05 g of this fraction was put into the reactor. The catalyst was first activated for 2 h at 623 K (heating rate 15 K min−1) under nitrogen flow (40 cm3 min−1). Subsequently, a 0.5 cm3 of HDN (Fluka, GC grade) was passed continuously into the catalyst at 623 K. The substrate was delivered with a pump system (KD Scientific) and vaporised before being passed through the catalyst with the flow of nitrogen carrier gas (40 cm3 min−1). The reaction products were collected for 30 min downstream of the reactor in a cold trap (liquid nitrogen and 2-propanol) and analyzed by gas chromatography (SRI 310 C, MXT-1 column 30 m, temperature of column 373 K) with a TCD detector. Helium was applied as a carrier gas.Dibutyl sulfide oxidation with H2O2
The catalytic reaction between dibutyl sulfide and hydrogen peroxide was carried out in a glass flask equipped with a water jacket (connected to the cryostat – Labortechnik Medingen K21), magnetic stirrer, a reflux condenser and a membrane for sampling. Before the reaction, 0.04 g of calcined catalyst was placed into the flask, where methanol (10 cm3) was added. After stirring for 1 h, 96% dibutyl sulfide (2 mmol – Aldrich) was added, followed by the dropwise addition of 35% hydrogen peroxide (2 mmol – Aldrich). The oxidation was conducted by stirring at 303 K for 8 h. The reaction mixture was analysed each 1 h using a gas chromatograph (Varian CP 3800) equipped with a 60 m VF-5ms capillary column, working in the temperature range of 333–523 K (temperature ramp 10 K min−1), and a FID detector.Results and discussion
Texture/structure characterization
Fig. 1 presents the X-ray patterns of the HMCM-22 and HMCM-56 materials. All synthesized zeolites show well-defined crystal structures. The appearance of XRD peaks characteristic of the materials with MWW structures indicates the successful synthesis of the MCM-22 and MCM-56 samples. Impregnation of both layered and delaminated MWW zeolites with Nb or Zr does not change the zeolites' structure (XRD patterns not shown here). No XRD reflections from crystalline metal oxide phases were observed in the XRD patterns. The absence of the peaks from metal oxides can be caused by good dispersion of metal oxide, or if the Nb and Zr are present not only in the form of crystalline oxides in the samples prepared.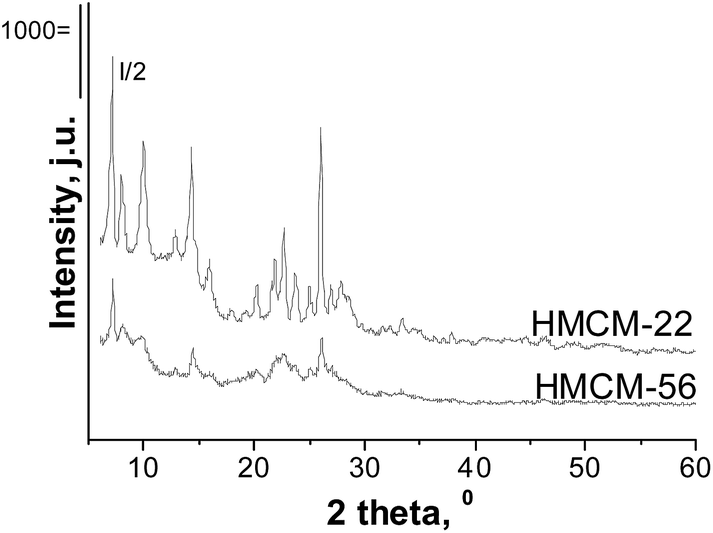 | ||
| Fig. 1 XRD patterns of HMCM-22 and HMCM-56. | ||
The materials prepared show surface areas and pore volumes (Table 1) typical of HMCM-22. In the case of the HMCM-56 zeolites some degradation or pore blockage seem to occur if compared with the typical MCM-56 materials reported in the literature. Impregnation of MWW zeolites by metals (Nb or Zr) leads to changes in the textural parameters. In all cases, the BET surface areas of HMCM-22 and delaminated HMCM-56 decrease after metal introduction. This fact is caused by a decrease in the external surface area, which could be a consequence of the Nb and Zr being located partially outside the zeolite pores.
| Catalyst | Stotal (m2 g−1) | Smicro (m2 g−1) | Sexter (m2 g−1) | Vtotal (cm3 g−1) | Vmicro (cm3 g−1) |
|---|---|---|---|---|---|
| HMCM-22 | 461 | 341 | 120 | 0.38 | 0.18 |
| Nb/HMCM-22 | 423 | 336 | 87 | 0.27 | 0.18 |
| Zr/HMCM-22 | 419 | 317 | 101 | 0.39 | 0.17 |
| HMCM-56 | 212 | 119 | 92 | 0.84 | 0.06 |
| Nb/HMCM-56 | 195 | 119 | 76 | 1.14 | 0.06 |
| Zr/HMCM-56 | 183 | 122 | 60 | 1.22 | 0.06 |
The state of metals
To examine the state of niobium and zirconium in the zeolites synthesized in detail, XPS and UV-vis spectroscopy were used. The UV-vis spectra of niobium containing zeolites (Fig. 2) are characterized by bands at ca. 220 and 250 nm, which are typical of tetrahedrally coordinated niobium(V) species in different surroundings.43,44 The octahedrally coordinated niobium species gives a characteristic adsorption band at ca. 330 nm. This band is not observed in the spectra of both the Nb/HMCM-22 and the delaminated Nb/HMCM-56 zeolites. | ||
| Fig. 2 UV-vis spectra of Nb/HMCM-22, Zr/HMCM-22, Nb/HMCM-56, and Zr/HMCM-56. | ||
The UV-vis spectra of the zirconium modified zeolites (Fig. 2), i.e. Zr/HMCM-22 and delaminated Zr/HMCM-56, show one band at 207 nm. According to the literature, this band can be assigned to the ligand to metal charge transfer (LMCT) from O2− to an isolated Zr4+ ion in a tetrahedral coordination.45,46
For further investigation of the niobium and zirconium states and localization in the prepared materials, XPS spectroscopy was applied. The results obtained for all samples, as well as for niobium(V) and zirconium(IV) bulk oxides, are collated in Table 2. The XPS study indicated binding energies (BE) typical of niobium +5 and zirconium +4. The binding energies of Nb 3d and Zr 3d located in the zeolites are shifted to higher values than in the case of bulk niobium and zirconium oxides. These results are noted for all catalysts, both MCM-22 and delaminated MCM-56. Higher values of BE can suggest different surroundings of niobium and zirconium in zeolites than those in bulk oxides.
| Catalyst | BE [eV] | |||
|---|---|---|---|---|
| Nb5+ | Zr4+ | |||
| 3d5/2 | 3d3/2 | 3d5/2 | 3d3/2 | |
| Nb2O5 | 207.10 | — | — | — |
| HMCM-22 | — | — | — | — |
| Nb/HMCM-22 | 208.60 | 211.40 | — | — |
| Zr/HMCM-22 | — | — | 184.07 | 186.40 |
| ZrO2 | — | — | 182.50 | — |
| HMCM-56 | — | — | — | — |
| Nb/HMCM-56 | 207.60 | 210.30 | — | — |
| Zr/HMCM-56 | — | — | 182.78 | 185.17 |
Acidity measurements
The surface properties of the prepared zeolites were investigated by infrared spectroscopy combined with the adsorption of pyridine. According to the literature, when pyridine is coordinated to Lewis acid sites (LAS), characteristic bands at ∼1450 cm−1 and ∼1610 cm−1 appear.47 The intensity of the first one is related to the number of LAS, whereas the position of the second band characterizes the strength of the LAS. Adsorption of pyridine on Brønsted acid sites (BAS) gives a band at ∼1550 cm−1 and two other ones in the 1620–1640 cm−1 range coming from the vibration of pyridinium cations.48 Moreover, pyridine can also interact with hydroxyls on the zeolite surface giving adsorption bands at 1596 cm−1 and 1445 cm−1 assigned to hydrogen bonded species. The IR band at ca. 1490 cm−1 is assigned to both pyridine coordinated to LAS and pyridinium cations.The spectra (normalized to 10 mg of catalyst) after pyridine adsorption at 473 K and outgassing at 573 K for 0.5 h are presented in Fig. 3. All prepared catalysts show acidic character. The characteristic bands from pyridine adsorbed on Lewis (1610, 1453 cm−1) and Brønsted (1544 and 1635 cm−1) acidic sites are well visible. The main difference between the layered and the delaminated materials is the intensity of these bands. All HMCM-22 and metal-modified zeolites exhibit more intense bands than the delaminated HMCM-56 and metal modified materials.
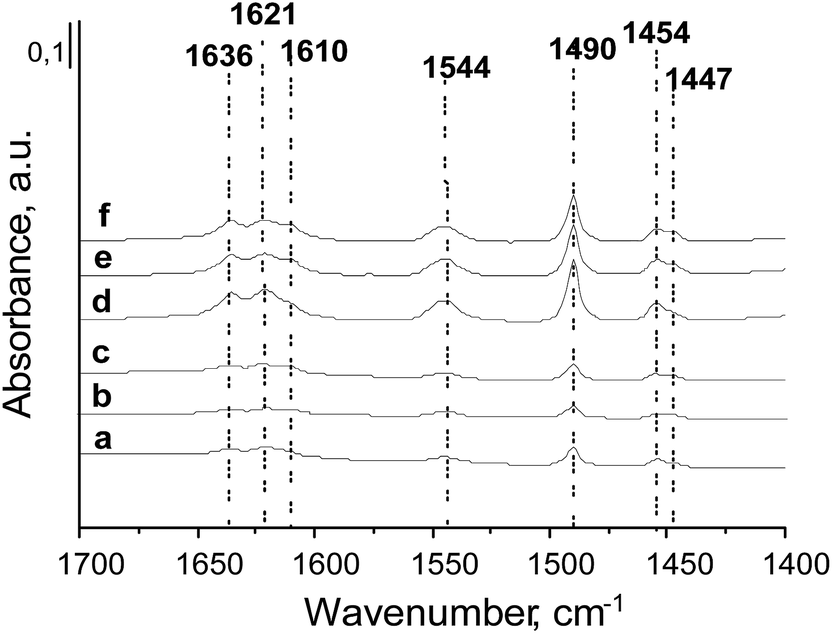 | ||
| Fig. 3 FTIR spectra after pyridine adsorption at 473 K on: (a) HMCM-56; (b) Zr/HMCM-56; (c) Nb/HMCM-56; (d) HMCM-22; (e) Zr/HMCM-22; and (f) Nb/HMCM-22. Spectra are normalized to 10 mg of zeolite. | ||
In consequence, the calculated numbers of BAS (1544 cm−1) and LAS (1453 cm−1) are higher for HMCM-22 and its metal modified materials (Table 3). The introduction of niobium and zirconium decreases the number of BAS and LAS. The drop in the LAS and BAS numbers (after evacuation at 573 K) oscillates between 22 and 35% independently of the zeolite type. The exception is Nb/HMCM-56 in which only a 6% drop in the LAS (after evacuation at 573 K) is observed after Nb loading. It suggests the higher strength of the LAS in this material. A similar impact of metal (Mo) introduction on HMCM-22 acidity had been observed before by Huang et al.,50 and also in our previous study with molybdenum.42
| Catalyst | Number of Lewis acid sites occupied by pyridine [μmol g−1] after evacuation at | Number of Brønsted acid sites occupied by pyridine [μmol g−1] after evacuation at | ||||
|---|---|---|---|---|---|---|
| 473 K | 523 K | 573 K | 473 K | 523 K | 573 K | |
| HMCM-22 | 1914 | 1651 | 1455 | 12961 |
12080 |
10895 |
| Nb/HMCM-22 | 1538 | 1164 | 1105 | 9216 | 9644 | 8663 |
| Zr/HMCM-22 | 1748 | 1380 | 1125 | 9750 | 8742 | 7786 |
| HMCM-56 | 837 | 679 | 538 | 3400 | 2722 | 2286 |
| Nb/HMCM-56 | 869 | 651 | 516 | 3009 | 2452 | 1725 |
| Zr/HMCM-56 | 748 | 565 | 413 | 3141 | 2330 | 1699 |
Fig. 4 presents the FTIR spectra in the hydroxyl region for HMCM-22 as an example of changes in this region after pyridine adsorption. According to the literature,51 the band at 3620 cm−1 corresponds to bridged hydroxyl groups (Al–OH–Si), whereas the band at 3670 cm−1 is often ascribed to hydroxyl groups linked to extra-framework aluminium species (generated upon calcination); and the bands at ∼3730 and 3748 cm−1 are assigned to internal and external silanol groups, respectively. Two IR bands dominate in all spectra, 3621 cm−1 (BAS) and 3748 cm−1 (originated from external silanol groups). Moreover, the 3728 cm−1 band coming from vibrations in the internal silanol groups as well as the bands at 3671 cm−1 assigned to hydroxyls linked to the extra-framework aluminium species are present in the spectra presented in Fig. 4. Introduction of metals (Nb or Zr) reduces the intensity of the 3620 cm−1 and 3748 cm−1 bands in the spectra of Nb/HMCM-22 or Zr/HMCM-22. These results indicate the interaction of metals species with BAS and external silanol groups. Interestingly, internal silanol groups and hydroxyls linked to extra-framework aluminium species are not involved in the modification of HMCM-22 with metal species.
 | ||
| Fig. 4 FTIR spectra after outgassing at 673 K for 2 h of: (a) HMCM-22; (b) Nb/HMCM-22; and (c) Zr/HMCM-22. Spectra are normalized to 10 mg of zeolite. | ||
For the evaluation of the acid/base properties of the synthesized zeolites in reaction conditions, dehydration and cyclisation of 2,5-hexanedione was carried out. The main products of this reaction are: 2,5-dimethylfuran (DMF) formed in the presence of acidic centers and 3-methyl-2-cyclopentenone (MCP) formed in the presence of basic centers.52,53 The ratio of MCP to DMF products allows the estimation of the acid-base character of the catalyst. If acidic centers dominate on the catalyst surface, the selectivity ratio MCP/DMF is <1. The ratio MCP/DMF > 1 indicates that basicity is dominant. When MCP/DMF ∼ 1, the catalyst has both acid and base characters.
The results of the dehydration and cyclisation of 2,5-hexanedione performed over niobium and zirconium catalysts are shown in Table 4. All tested zeolites show acidic character, and the main product observed is DMF, which is formed on Brønsted acid sites. It is important to point out that for the MCM-22 type zeolites (Nb/HMCM-22 and Zr/HMCM-22) a higher value of 2,5-HDN conversion was observed. The higher conversion on Nb/HMCM-22 and Zr/HMCM-22 than on the appropriate MCM-56 zeolites, should be related to the higher number of BAS proved by pyridine adsorption. However, not only the number of BAS influences the activity of catalyst, but also the nature of the metal. Zirconium containing catalysts show almost two times lower conversion of 2,5-HDN than niobium catalysts.
| Catalyst | Conversion of 2,5-HDN, % | Selectivity, % | MCP/DMF | |
|---|---|---|---|---|
| MCP | DMF | |||
| Nb/HMCM-22 | 88 | 2 | 98 | 0.02 |
| Zr/HMCM-22 | 48 | 18 | 82 | 0.21 |
| Nb/HMCM-56 | 74 | 4 | 96 | 0.04 |
| Zr/HMCM-56 | 34 | 23 | 77 | 0.29 |
Dibutyl sulfide oxidation with H2O2
Fig. 5 presents dibutyl sulfide conversion versus the reaction time on niobium and zirconium catalysts. Dibutyl sulfide oxidation with hydrogen peroxide performed in the absence of a catalyst led to sulfide conversion of 78% after 8 h. The application of catalysts significantly increases the reaction rate, leading to a final conversion of 87% (Nb/HMCM-22) and 95% (Nb/HMCM-56) after much shorter times and a very high yield of sulfoxide. The modification of zeolites with zirconium does not improve the activity of catalysts used. The reaction in the presence of both Zr/HMCM-22 and delaminated Zr/HMCM-56, proceeds very slowly, and the obtained conversions are similar (Zr/HMCM-56) or even lower (Zr/HMCM-22) than in the case of oxidation of dibutyl sulfide without a catalyst. The low activity of zirconium containing zeolites can be caused by the strong interaction between zirconium species in the catalyst and sulfur containing compounds. Similar properties of this transition metal were observed in the papers54,55 for the interaction between SO2 and H2S with ZrO2. A totally different effect is observed in the presence of both Nb/HMCM-22 and Nb/HMCM-56 samples, which led to the faster reaction than in the blank test (Fig. 5). After 10 min of the reaction time the most active catalyst, Nb/HMCM-56, led to 80% conversion of dibutyl sulfide (with a very high yield of sulfoxide – Fig. 6). The high activity of niobium containing materials can be explained by the ability of niobium to activate the hydrogen peroxide.34 It has been postulated that the electrophilicity of oxygen in H2O2 increases in contact with an oxometal group,56 thus the nucleophilic attack of sulfur is more favourable. | ||
| Fig. 5 Conversion vs. reaction time in dibutyl sulfide oxidation with H2O2 (mass of catalyst – 40 mg; reaction temperature – 303 K; methanol – 10 mL; dibutyl sulfide – 2 mmol; H2O2 – 2 mmol). | ||
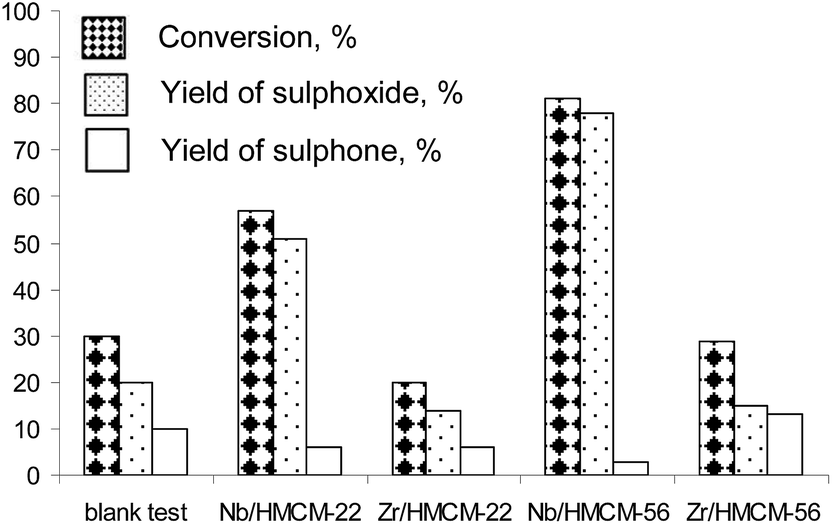 | ||
| Fig. 6 Conversion and yield to sulfoxide and sulfone in dibutyl sulfide oxidation with H2O2 after 10 min of the reaction (mass of catalyst – 40 mg; reaction temperature – 303 K; methanol – 10 mL; dibutyl sulfide – 2 mmol; H2O2 – 2 mmol). | ||
In spite of the rapid reaction progress for niobium containing zeolites, it can be observed that Nb/HMCM-56 is more active than Nb/HMCM-22. This feature could be related to the different structure of both materials (e.g. volume of mesopores).
Catalysts used in this work exhibit Brønsted (BAS) and Lewis (LAS) acid sites. Both of them can participate in the oxidation of dibutyl sulfide. To consider the role of these centres, one has to discuss the chemisorption of both reagents. Dibutyl sulfide is expected to chemisorb, if at all, on niobium or zirconium cations. Recently, it was established33 that the interaction of the acidic surface hydroxyls with hydrogen peroxide gives rise to the formation of hydrosuperoxo species (HO2−). These species interact with the excess of H2O2 towards hydroxyl radicals, which are the most active species in the catalytic oxidations. The role of the niobium centres is to trap superoxo (O2−˙) and peroxo (O22−) species on the surface. This process leads to increased production of hydroxyl radicals (˙OH). They are generated via electroprotic liquid phase reaction H2O2(aq) + HO2−(aq) = ˙OH(aq) + O2−˙(aq) + H2O(aq). Trapping of superoxo and peroxo species by niobium centres leads to a substantial shift of the reaction equilibrium to the right enhancing the production of hydroxyl radicals, which interact with dibutyl sulfide very quickly. This process is responsible for the substantial increase of oxidation activity at the beginning of the reaction when the concentration of H2O2 is the highest. If the concentration of BAS on the catalyst surface is higher, as in the MCM-22 series, higher production of hydroxyl radicals is expected and it can lead to the easy recombination of radicals decreasing the catalytic activity. In the case of Zr containing catalysts dibutyl sulfide seems to be preferentially chemisorbed on zirconium LAS. It is known that ZrO2 strongly chemisorbs sulfur compounds,54,55 and also dialkyl sulfides.57 The strongly held dibutyl sulfide is not reactive towards hydrogen peroxide and Zr species are blocked. Therefore, Zr containing catalysts are not active (the activity is lower than in the blank test).
The discussion presented above allows us to suggest that the effective reaction pathway in the oxidation of dibutyl sulfide proceeds through the generation of hydroxyl radicals interacting with dibutyl sulfide from the liquid phase. One can find confirmation of the interaction of the Nb containing zeolites with H2O2 in the UV-vis spectra after treatment of the catalysts with hydrogen peroxide. The example for Nb/HMCM-22 is shown in Fig. 7. Zeolite treated with hydrogen peroxide showed an increase in the absorption intensity in the range 300–400 nm, assigned to charge transfer from O2−˙ (superoxo species) to niobium.33,34
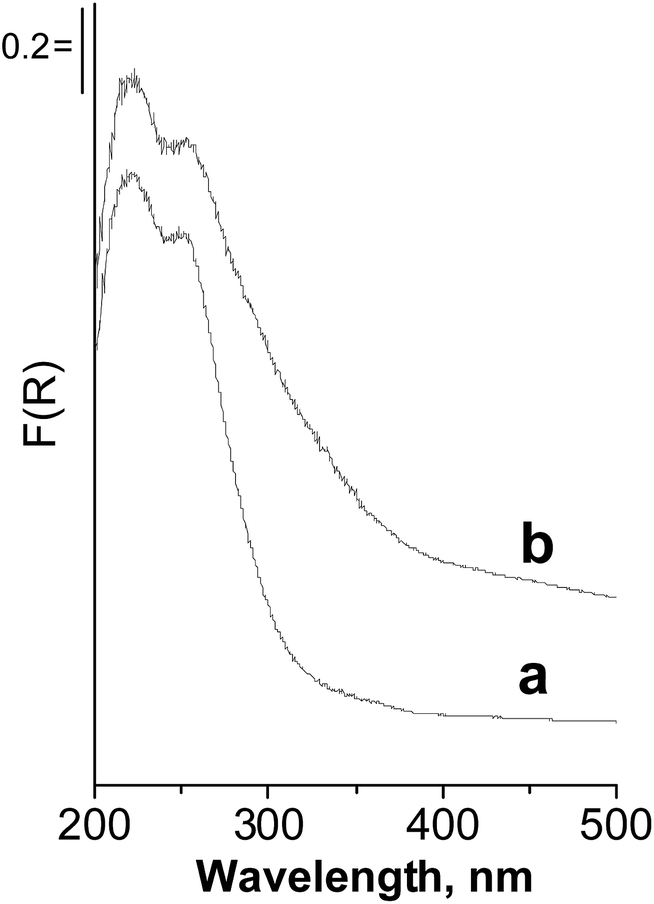 | ||
| Fig. 7 UV-vis spectra of: (a) Nb/HMCM-22, (b) Nb/HMCM-22 after treatment with hydrogen peroxide. | ||
The catalysts after the reaction were studied by XPS and UV-vis methods. The XPS results showed the same concentration of metal species before and after oxidation of dibutyl sulfide. It suggested that leaching of the metal phase does not occur. The UV-vis spectra of catalysts after the reaction reveal some increase of the band at ca. 220 nm caused by the loading of sulfur compounds in the pores.
The comparison of the results presented in this paper and those published for Mo containing zeolites42 allows us to present the following order of activity dependence on the nature of the metal modifier: Mo > Nb ≫ Zr.
The oxidation of dibutyl sulfide with hydrogen peroxide showed that the zeolite structure (MCM-22 or MCM-56) and the nature of the modifier (Nb or Zr) have an impact on the catalytic behaviour. MCM-56 modified with niobium gives very high activity and selectivity for sulfoxide.
Conclusions
• Modification of HMCM-22 and HMCM-56 zeolites with niobium and zirconium does not change the structure of catalysts, as confirmed by XRD study.• Crystalline metal oxides are not detected on the zeolite surfaces and it has been proved that both niobium and zirconium species are present in a tetrahedral coordination.
• Metal loading involves the interaction with BAS and external silanol groups on the zeolite surface. Therefore, modification of both HMCM-22 and HMCM-56 zeolites with Nb or Zr decreases the number of BAS.
• Nb species and the texture of zeolite (MCM-22 vs. MCM-56) play a crucial role in the oxidation of sulfide to sulfoxide (>90% conversion of dibutyl sulfide is reached after 1 h of the reaction on Nb/HMCM-56).
• Zirconium loaded on zeolites even decreases the rate of the reaction performed without the use of any catalyst due to its strong interaction with sulfur compounds.
Acknowledgements
We are grateful to the National Science Center in Poland for support (project no. 2011/01/B/ST5/00847). Acknowledgements to Andrzej Nowak and Dr Maciej Trejda for taking part in some experimental works.Notes and references
- Zeolites and Catalysis: Synthesis Reactions and Applications, ed. J. Cejka, A. Corma and S. I. Zones, Wiley-VCH, Weinheim, vol. 1–2, 2010 Search PubMed.
- M. K. Rubin and P. Chu, U.S. Patent, 4,954,325, 1990.
- M. E. Leonowicz, J. A. Lawton, S. L. Lawton and M. K. Rubin, Science, 1994, 264, 1910 CAS.
- A. Corma, C. Corell and J. Perez-Pariente, Zeolites, 1995, 15, 2 CrossRef CAS.
- W. J. Roth and D. L. Dorset, Microporous Mesoporous Mater., 2011, 142, 32 CrossRef CAS PubMed.
- W. J. Roth and J. Cejka, Catal. Sci. Technol., 2011, 1, 43 CAS.
- A. S. Fung, S. L. Lawton and J. Roth, US Patent 5,362,697, 1994.
- G. Bellusi, G. Perego, M. G. Cierici and A. Giusi, European Patent, 293032, 1988.
- S. I. Zones, European Patent, 231860, 1987.
- L. Puppe and J. Weisser, US Patent 4,439,409, 1984.
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo, Nature, 2009, 461, 246 CrossRef CAS PubMed.
- A. Corma, V. Fornes and S. B. C. Pergher, World Patent, WO9717290A1, 1997.
- A. Corma, V. Fornes, S. B. C. Pergher, T. L. M. Maesen and J. G. Buglass, Nature, 1998, 396, 353 CrossRef CAS PubMed.
- W. J. Roth, C. T. Kresge, J. C. Vartuli, M. E. Leonowicz, A. S. Fung and S. B. McCullen, Catalysis by Microporous Materials, Stud. Surf. Sci. Catal., ed. H. K. Beyer, H. G. Karge, I. Kiricsi and J. B. Nagy, Elsevier, New York, vol. 94, 1995, p. 301 Search PubMed.
- U. Diaz, ISRN Chem. Eng., 2012, 2012, 1 CrossRef PubMed.
- W. J. Roth, P. Nachtigall, R. E. Morris and J. Cejka, Chem. Rev., 2014, 114, 4807 CrossRef CAS PubMed.
- U. Diaz and A. Corma, Dalton Trans., 2014, 43, 10292 RSC.
- T. Selvam, A. Inayat and W. Schwieger, Dalton Trans., 2014, 43, 10365 RSC.
- B. Marler and H. Gies, Eur. J. Mineral., 2012, 24, 405 CrossRef CAS.
- W. J. Roth, B. Gil and B. Marszalek, Catal. Today, 2014, 227, 9 CrossRef CAS PubMed.
- G. G. Juttu and R. F. Lobo, Microporous Mesoporous Mater., 2000, 40, 9–23 CrossRef CAS.
- S. L. Lawton, M. E. Leonowicz, R. D. Partridge, P. Chu and M. K. Rubin, Microporous Mesoporous Mater., 1998, 23, 109 CrossRef CAS.
- Y. Wang, Y. M. Liu, L. L. Wang, H. H. Wu, X. H. Li, M. Y. He and P. Wu, J. Phys. Chem. C, 2009, 113, 18753 CAS.
- W. J. Roth, Stud. Surf. Sci. Catal., 2005, 158, 19 CrossRef.
- B. Gil, W. Makowski, B. Marszalek, W. J. Roth, M. Kubu, J. Cejka and Z. Olejniczak, Dalton Trans., 2014, 43, 10501 RSC.
- F. S. O. Ramos, M. K. De Pietre and H. O. Pastore, RSC Adv., 2013, 7, 2084 RSC.
- P. Wu, T. Tatsumi, T. Komatsu and T. Yashima, Chem. Lett., 2000, 29, 774 CrossRef.
- J. Ruan, P. Wu, B. Slater and O. Terasaki, Angew. Chem., 2005, 44, 6719 CrossRef CAS PubMed.
- S. J. Kim, K. D. Jung and O. S. Joo, J. Porous Mater., 2004, 11, 211 CrossRef CAS.
- A. A. Teixeira-Neto, L. Marchese, G. Landi, L. Lisi and H. O. Pastore, Catal. Today, 2008, 1, 133 Search PubMed.
- Y. Wu, J. Wang, P. Liu, W. Zhang, J. Gu and X. Wang, J. Am. Chem. Soc., 2010, 132, 17989 CrossRef CAS PubMed.
- W. J. Roth, W. Makowski, B. Marszalek, P. Michorczyk, W. Skuza and B. Gil, J. Mater. Chem. A, 2014, 2, 15722 CAS.
- M. Ziolek, I. Sobczak, P. Decyk, K. Sobanska, P. Pietrzyk and Z. Sojka, Appl. Catal., B, 2015, 164, 288 CrossRef CAS PubMed.
- M. Ziolek, I. Sobczak, P. Decyk and L. Wolski, Chem. Commun., 2013, 37, 85 CAS.
- O. Saur, T. Chevreaur, J. Lamotte, J. Travert and J.-C. Lavalley, J. Chem. Soc., Faraday Trans. 1, 1981, 27, 427 RSC.
- J. A. Melero, J. A. Melero, L. F. Bautista, J. Iglesias, G. Morales and R. Sánchez-Vázquez, Catal. Today, 2012, 195, 44 CrossRef CAS PubMed.
- K. K. Shah, J. Saikia, D. Saikia and A. K. Talukdar, Mater. Chem. Phys., 2012, 134, 43 CrossRef CAS PubMed.
- I. Nowak and M. Ziolek, Chem. Rev., 1999, 99, 3603 CrossRef CAS PubMed.
- E. I. Ko, R. Bafrali, N. T. Nuhfer and N. J. Wagner, J. Catal., 1985, 95, 260 CrossRef CAS.
- G. Deo and I. E. Wachs, J. Catal., 1991, 129, 307 CrossRef CAS.
- M. Ziolek, Catal. Today, 2003, 78, 47 CrossRef CAS.
- A. Wojtaszek-Gurdak, M. Trejda, D. Kryszak and M. Ziolek, Microporous Mesoporous Mater., 2014, 197, 185 CrossRef CAS PubMed.
- D. Blasco-Jiméneza, I. Sobczak, M. Ziolek, A. J. López-Peinado and R. M. Martín-Aranda, Catal. Today, 2010, 152, 119 CrossRef PubMed.
- B. Kilos, A. Tuel and M. Ziolek, Catal. Today, 2006, 118, 416 CrossRef CAS PubMed.
- E. Rodriguez-Castellon, A. Jimenez-Lopez, P. Maireles-Torres, D. J. Jones, J. Roziere, M. Trombetta, G. Busca, M. Lenarda and L. Storaro, J. Solid State Chem., 2003, 175, 159 CrossRef CAS.
- J. Goscianska, M. Ziolek, E. Gibson and M. Daturi, Catal. Today, 2010, 152, 33 CrossRef CAS PubMed.
- M. Ziolek, I. Nowak and J. C. Lavalley, Catal. Lett., 1997, 45, 259 CrossRef CAS.
- S. Khabtou, T. Chevreau and J. C. Lavalley, Microporous Mater., 1994, 3, 133 CrossRef CAS.
- B. Gil, B. Marszałek, A. Micek-Ilnicka and Z. Olejniczak, Top. Catal., 2010, 53, 1340 CrossRef CAS.
- S.-J. Huang, Q. Zhao, W.-H. Chen, X. Han, X. Bao, P.-S. Lo, H.-K. Lee and S.-B. Liu, Catal. Today, 2004, 97, 25 CrossRef CAS PubMed.
- P. Matias, J. M. Lopes, S. Laforge, P. Magnoux, P. A. Russo, M. M. L. Ribeiro Carrott, M. Guisnet and F. Ramoa Ribeiro, J. Catal., 2008, 259, 190 CrossRef CAS PubMed.
- R. M. Dessau, Zeolites, 1990, 10, 205 CrossRef CAS.
- J. J. Alcaraz, B. J. Arena, R. D. Gillespie and J. S. Holmgren, Catal. Today, 1998, 43, 89 CrossRef CAS.
- M. Ziolek, J. Kujawa, O. Saur and J. C. Lavalley, J. Mol. Catal. A: Chem., 1995, 97, 49 CrossRef CAS.
- M. Ziolek, J. Kujawa, O. Saur, A. Aboulayt and J. C. Lavalley, J. Mol. Catal. A: Chem., 1996, 112, 125 CrossRef CAS.
- A. L. Maciuca, C. E. Ciocan, E. Dumitriu, F. Fajula and V. Hulea, Catal. Today, 2008, 138, 33 CrossRef CAS PubMed.
- M. Ziolek, O. Saur, J. Lamotte and J.-C. Lavalley, J. Chem. Soc., Faraday Trans., 1994, 90, 1029 RSC.
| This journal is © The Royal Society of Chemistry 2015 |