
Catalytic hydrogenation of phenol, cresol and guaiacol over physically mixed catalysts of Pd/C and zeolite solid acids†
Abstract
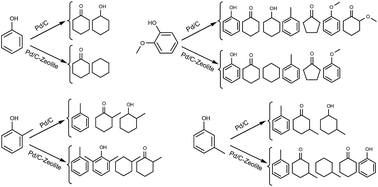
Highly reactive phenolic compounds of pyrolysis bio-oil are recognized as a major cause of the unpleasant properties of this biofuel. Catalytic hydrodeoxygenation of phenolic compounds of bio-oil is an efficient technique for improving the quality of bio-oil. Dual function catalysts consisting of metal and acid sites are usually used for transformation of bio-oil/bio-oil model compounds to high value hydrocarbons. Metal and acid sites are generally involved in hydrogenation/hydrodeoxygenation and dehydration/hydrocracking/dealkylation/alkylation reaction mechanisms, respectively. In this work, the product selectivity of hydrogenation of phenol, o-cresol, m-cresol and guaiacol was investigated over combined catalysts of Pd/C with zeolite solid acids of HZSM-5 (Si/Al of 30, 50 and 80) and HY (Si/Al of 30 and 60). Catalytic activity and product distribution in the hydrogenation process were affected by the density and strength of zeolite acid sites. HZSM-5 (30) with only weak acid sites showed lower cyclohexane selectivity compared with HZSM-5 (50) and HZSM-5 (80) which had both weak and strong acid sites. HY (30) and HY (60) containing only strong acid sites favored production of cycloketones.
 Please wait while we load your content...
Please wait while we load your content...

