
Synthesis and properties of a bio-based epoxy resin from 2,5-furandicarboxylic acid (FDCA)
Jun Deng,
Xiaoqing Liu*,
Chao Li,
Yanhua Jiang and
Jin Zhu
Ningbo Key Laboratory of Polymer Materials, Ningbo Institute of Material Technology and Engineering, Chinese Academy of Sciences, Ningbo 315201, China. E-mail: liuxq@nimte.ac.cn; Fax: +86-574-86685925
First published on 28th January 2015
Abstract
A bio-based epoxy monomer, diglycidyl ester of 2,5-furandicarboxylic acid (DGF) was synthesized for the first time from the renewable 2,5-furandicarboxylic acid (FDCA). For comparison study, its petroleum-based counterpart, diglycidyl ester of terephthalic acid (DGT) was also prepared. Their chemical structures were confirmed in detail by 1H NMR, 13C NMR and FT-IR before they were cured by methylhexahydrophthalic anhydride (MHHPA) and poly(propylene glycol)bis(2-aminopropyl ether) (D230), respectively. The curing behaviors were investigated using differential scanning calorimetry (DSC). The thermal mechanical properties and thermal stabilities of the cured resins were evaluated using dynamic mechanical analysis (DMA) and thermogravimetric analysis (TGA). Results showed that DGF displayed higher curing activity, elevated glass transition temperature and similar mechanical properties compared with those of the cured DGT. This study indicated that FDCA had a huge potential to replace the petroleum-based terephthalic acid in the synthesis of epoxy resins with satisfactory performance.
1 Introduction
The conversion of biomass into useful polymeric materials has been regarded as one of the effective methods to save fossil resources and protect our living environment. According to the origin and production of bio-based polymers, it could be divided into three main categories: natural polymers, synthesized bio-based polymers and those produced by microorganisms or bacteria.1 Undoubtedly, the synthesized bio-based polymers are most popular and promising. A large quantity of polyesters,2–7 such as polylactic acid (PLA), poly(hydroxyalkanoates) (PHAs), poly(butylene succinate) (PBS) and polyethylene (PE) has been synthesized from biomass and commercialized successfully already. However, compared with the well-known engineering plastics, such as poly(ethylene terephthalate) (PET), polycarbonate (PC) and diglycidyl ether of biphenyl A (DGEBA), their mechanical and thermal properties are still subjects to be improved upon for the applications of structural or engineering materials. Based on the chemical structure comparison in Scheme 1, the lack of aromatic or rigid ring structure in current bio-based polymers' architectures might be the reason for their relatively weak properties. Therefore, the exploration of aromatic or rigid compounds derived from renewable resources should be significant for the next generation of bio-based polymers.8,9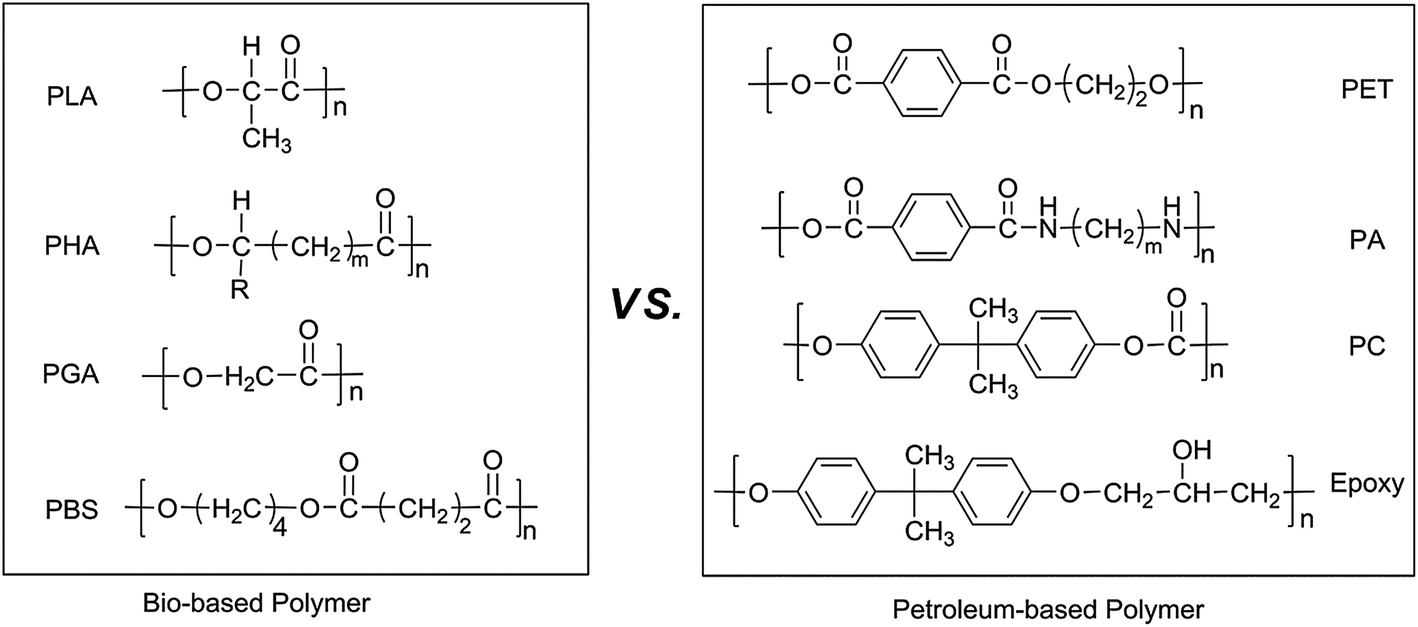 | ||
| Scheme 1 Chemical structures of current popular bio-based polymers and petroleum-based engineering plastics. | ||
2,5-Furandicarboxylic acid (FDCA), which can be derived from polysaccharides or sugars through the oxidation of hydroxymethylfurfural (HMF),10,11 has been identified as one of top 12 potential platform molecules likely to play an important role in establishing the green chemistry industry by the US department of Energy.12 Due to its structure similarity to that of terephthalic acid (TPA) and rapid progress on large scale production technology, FDCA might be one of the most promising bio-based compounds for TPA substitute. In fact, more and more explorations on the synthesis of polyester,13–17 polyamide18–20 and polyurethane21–24 containing FDCA units have been reported. FDCA has shown a huge potential in the synthesis of bio-based plastics with enhanced properties. For instance, including the comparable or even better thermo-mechanical properties, the barrier properties with regard to oxygen and carbon dioxide permeability of poly(ethylene 2,5-furandicarboxylate) (PEF) were much better than those of PET.25
Epoxy resin has been widely used in the field of aerospace, coatings, adhesives and composites due to its excellent properties. Nowadays, the dominant commercial epoxy resin, diglycidyl ether of bisphenol A (DGEBA) was produced completely from fossil resource and the toxicity associated with bisphenol A has been recognized extensively.26 In addition, compared with the activities and achievements on bio-based thermoplastics, researches on the subject of bio-based thermosetting resins need much more attention. Considering the concept of bio-based polymers and their development tendency, the thermosetting resins derived from biomass, especially the high-performance ones, should also have a bright future.
Up to now, several bio-based recourse, such as plant oils,27–33 lignin,34,35 rosin,36–39 starch,40 sucrose,41 gallic acid42,43 and isosorbide44 have been employed to prepare the bio-based epoxies.45–47 However, the resulting epoxy resins usually exhibited lower glass transition temperature,48 poor mechanical properties,31 slow curing reaction or unstable properties49 due to the structures' defects of the bio-based compounds. For example, the epoxy derived from plant oil or fatty acid usually showed poor mechanical and thermal properties because of its long soft aliphatic chain.50 How to stabilize the properties of lignin-based epoxy is still a challenge in front of us. The severe brittleness of epoxy derived from gallic acid and rosin51,52 should gain much more attention. Apparently, the platform building blocks selection and structure design are key factors to determine the properties of bio-based synthetic epoxy resins.
Based on its unique structures and encouraging performance in the synthesis of bio-based thermoplastics, the utilization of FDCA might be a promising start to the bio-based epoxy with satisfied properties. In this study, diglycidyl ester of 2,5-furandicarboxylic acid (DGF) and its petroleum-based counterpart diglycidyl ester of terephthalic acid (DGT) (Scheme 2) were synthesized. In order to compare their performance and study the possibility of FDCA for high performance epoxy synthesis, their curing reactions with MHHPA and D230 as well as the properties of the cured resins in terms of thermal stabilities, mechanical and thermal properties were investigated.
 | ||
| Scheme 2 Chemical structures of DGT, DGF, MHHPA and D230. | ||
2 Experimental
2.1 Materials and reagents
2,5-Furandicarboxylic acid was purchased from Sichuan Dagaote Technology Co., Ltd, China. Allyl bromide, m-chloroperoxybenzoic acid (m-CPBA) (85%) and poly(propylene glycol)bis(2-aminopropyl ether) D230 were obtained from Aladdin Reagent, China. 2-Ethyl-4-methylimidazole, methylhexahydrophthalic anhydride (MHHPA), epichlorohydrin (ECH) and terephthalic acid (TPA) were obtained from Sinopharm Chemical Reagent Co., Ltd, China. All the chemicals were used as received.2.2 Preparation of bis(prop-2-enyl)furan-2,5-dicarboxylate (FDCE)
FDCA (50 g, 0.32 mol), N,N-dimethylformamide (DMF, 140 mL), allyl bromide (143 g, 1.18 mol), acetone (150 mL), triethylamine (73 g, 0.72 mol) were placed in a round-bottomed flask equipped with a magnetic stirrer, a thermometer and a reflux condenser. The mixture was heated and refluxed for 24 h after the solid was dissolved completely in the solution. Then the salt precipitate was removed via filtration before the acetone, unreacted allyl bromide and triethylamine were removed from the filtrate by a rotary evaporator. In order to get rid of DMF in the raw product, the solution was washed by ionized water several times after it was diluted with dichloromethane. After that, dichloromethane was removed before it was dried with anhydrous magnesium sulphate (MgSO4). At last, a yellow solid product weighing (62 g, 82%) was obtained. The synthetic scheme is presented in Scheme 3.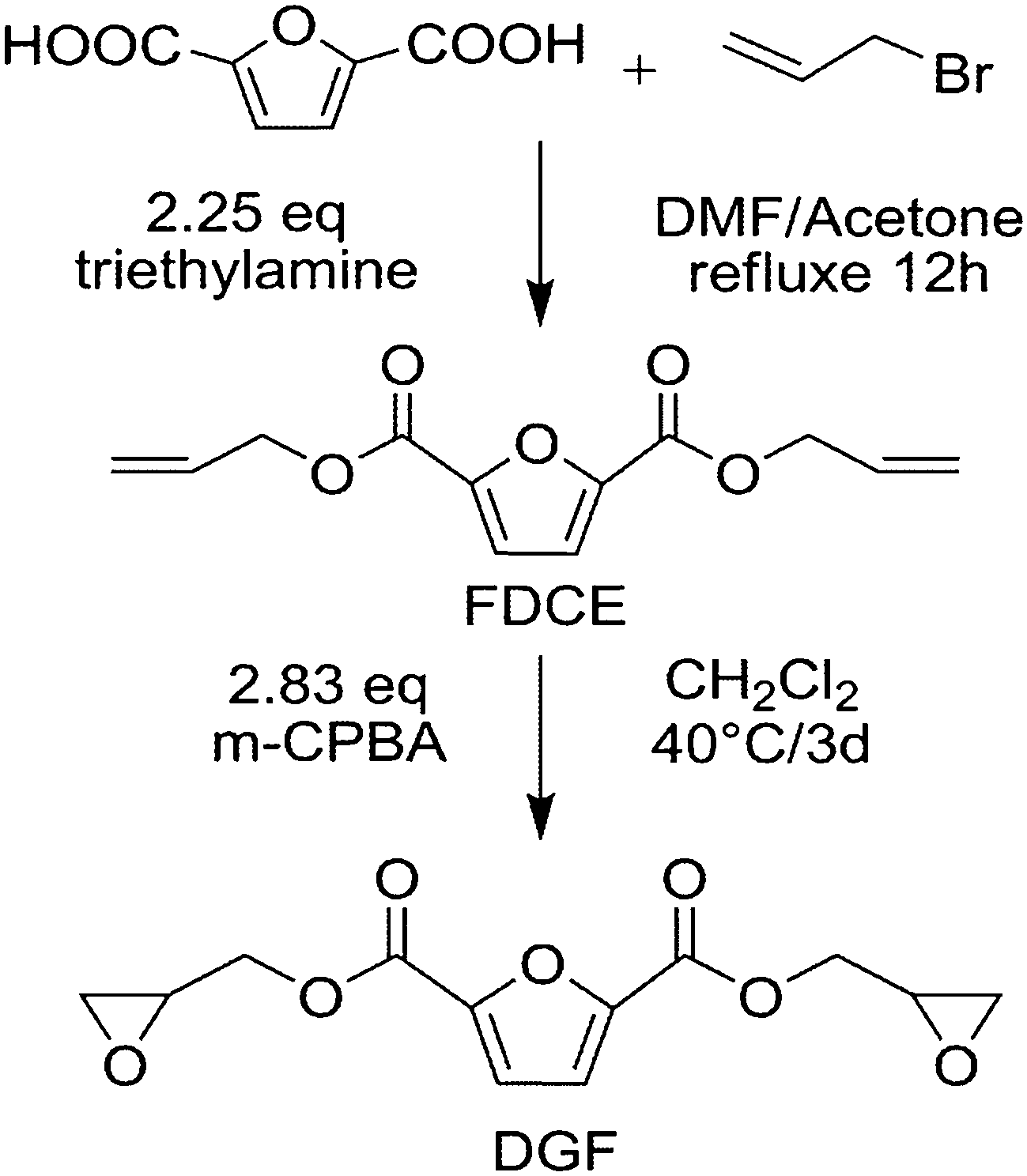 | ||
| Scheme 3 Synthesis procedure of DGF from FDCA. | ||
FT-IR (KBr, cm−1): 3085, 979 and 931 cm−1 (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 2946 cm−1 (–CH2–), 1713 cm−1 (CO), 1650 cm−1 (–CC–), 1287 cm−1 (–C–O–), 1578, 1223, 1018, 979, 825, and 765 cm−1 (furan ring CO and –C–O–). 1H NMR (400 MHz, acetone-d6, δ, ppm): 7.37 (s, 2H), 6.06 (m, J1 = 22.7 Hz, J2 = 10.9 Hz, J3 = 5.72 Hz, 2H), 5.44 (m, 2H), 5.30 (d, J = 10.5 Hz, 2H), 4.84 (d, J = 5.6 Hz, 4H). 13C NMR (400 MHz, acetone-d6, δ, ppm): 157.2 (2C), 146.7 (2C), 132.1 (2C), 118.6 (2C), 118.1 (2C), 65.5 (2C).
CH2), 2946 cm−1 (–CH2–), 1713 cm−1 (CO), 1650 cm−1 (–CC–), 1287 cm−1 (–C–O–), 1578, 1223, 1018, 979, 825, and 765 cm−1 (furan ring CO and –C–O–). 1H NMR (400 MHz, acetone-d6, δ, ppm): 7.37 (s, 2H), 6.06 (m, J1 = 22.7 Hz, J2 = 10.9 Hz, J3 = 5.72 Hz, 2H), 5.44 (m, 2H), 5.30 (d, J = 10.5 Hz, 2H), 4.84 (d, J = 5.6 Hz, 4H). 13C NMR (400 MHz, acetone-d6, δ, ppm): 157.2 (2C), 146.7 (2C), 132.1 (2C), 118.6 (2C), 118.1 (2C), 65.5 (2C).
2.3 Preparation of DGF
A three-necked round-bottomed flask equipped with a mechanical stirrer, a thermometer and a reflux condenser was charged with a solution of FDCE (60 g, 0.25 mol), dichloromethane (450 mL) and m-CPBA (110 g, 0.63 mol). The reactants were mixed and kept at 40 °C for 3 days. After that, the mixture was filtered and washed with a solution of 10% sodium sulfite followed by 10% sodium carbonate and distilled water. The organic part was dried with anhydrous MgSO4 and concentrated by a rotary evaporator. The raw product was washed by ether several times until it became white. The resultant product weighting (50 g, 72%) was obtained after recrystallization. The synthetic route is shown in Scheme 3.FT-IR (KBr, cm−1): 3151 and 3109 cm−1 (furan ring C–H), 3013 cm−1 (oxirane ring C–H), 2949 cm−1 (–O–CH2–), 1722 cm−1 (CO), 1575, 1447 and 1343 cm−1 (furan ring CC and its breaking absorption), 1293 cm−1 (ester C–O), 1267 and 1075 cm−1 (furan ring C–O), 1232 and 866 cm−1 (oxirane group C–O–C), 909 cm−1 (the breathing absorption of oxirane ring), 986, 815 and 766 cm−1 (2,5-disubstituted in furan ring). 1H NMR (400 MHz, CDCl3, δ, ppm): 7.27 (s, 2H), 4.67 (d, J = 3.2 Hz, H), 4.64 (d, J = 3.2 Hz, H), 4.20 (m, J1 = 12.2 Hz, J2 = 6.3 Hz, 2H), 3.34 (m, J1 = 6.4 Hz, J2 = 3.1 Hz, 2H), 2.90 (t, J = 4.5 Hz, 2H), 2.73 (m, J1 = 18.1 Hz, J2 = 9.0 Hz, 2H); 13C NMR (400 MHz, CDCl3, δ, ppm): 157.5 (2C), 146.5 (2C), 118.9 (2C), 65.9 (2C), 49.0 (2C), 44.8 (2C).
2.4 Preparation of DGT
TPA (66.4 g, 0.4 mol), epichlorohydrin (ECH, 600 g, 6.48 mol) and cetyl trimethyl ammonium chloride (CTMAC, 3.32 g, 0.01 mol) were added into a three-necked round-bottomed flask equipped with a magnetic stirrer, a thermometer and a reflux condenser. With continuous stirring, the mixture was heated to 90 °C and maintained at this temperature for 3 hours. Then, the reaction was cooled to 30 °C and 100 g aqueous solution of sodium hydroxide (40 wt%, 1 mol) was added drop by drop. After that, the reaction was continued at 30 °C for another 6 h before it was washed with deionized water five times. The organic layer was dried with anhydrous MgSO4 and concentrated. At last, the white powder weighting (90 g, 80%) was obtained.FT-IR (KBr, cm−1): 3063 cm−1 (benzene ring C–H), 3010 cm−1 (oxirane ring C–H), 2959 and 2930 cm−1 (oxirane ring C–H), 1716 cm−1 (CO), 1580 and 1500 cm−1 (benzene ring CC), 1280 cm−1 (ester C–O), 1250 and 1073 cm−1 (oxirane ring C–O–C), 1115 and 980 cm−1 (benzene ring C–H), 897 cm−1 (the breathing absorption of oxirane ring), 864 cm−1 (oxirane ring C–O–C), 829 (benzene ring 1,4-disubstituted C–H). 1H NMR (400 Hz, CDCl3-d6, δ, ppm) 8.16 (d, J = 20.8 Hz, 4H), 4.72 (d, J = 3.0 Hz, 2H), 4.70 (d, J = 3.0 Hz, 2H), 4.20 (m, J1 = 12.3 Hz, J2 = 6.4 Hz, 2H), 3.38 (m, J1 = 6.5 Hz, J2 = 2.9 Hz, 2H), 2.93 (t, J = 4.5 Hz, 2H), 2.76 (m, J1 = 4.8 Hz, J2 = 2.6 Hz, 2H); 13C NMR (400 MHz, CDCl3-d6, δ, ppm): 165.4 (2C), 133.7 (2C), 129.7 (4C), 65.9 (2C), 49.3 (2C), 44.7 (2C).
2.5 Curing procedure
DGF or DGT was cured with MHHPA in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 equivalent ratio, together with 2-ethyl-4-methylimidazole (0.5 wt% on the basis of the total weight of curing agent and epoxy). A small amount of dichloromethane was added to dissolve the solid epoxy and MHHPA powder before it was stirred for 30 min at room temperature to get a homogeneous system. The mixture was placed in a vacuum oven at 30 °C for 2 h to get rid of the solvent and then it was transferred to a mold with uniform cavity size before curing at 120 °C for 2 h, 150 °C for 2 h and 180 °C for 2 h. DGF or DGT was formulated with D230 in the same conditions, mixed in a 1:1 equivalent ratio. The mixture was cured at 100 °C for 2 h, 130 °C for 2 h and 150 °C for 2 h to achieve fully cured polymers. All the samples were cured under the same conditions and carefully removed from the mold for properties study.
:1 equivalent ratio, together with 2-ethyl-4-methylimidazole (0.5 wt% on the basis of the total weight of curing agent and epoxy). A small amount of dichloromethane was added to dissolve the solid epoxy and MHHPA powder before it was stirred for 30 min at room temperature to get a homogeneous system. The mixture was placed in a vacuum oven at 30 °C for 2 h to get rid of the solvent and then it was transferred to a mold with uniform cavity size before curing at 120 °C for 2 h, 150 °C for 2 h and 180 °C for 2 h. DGF or DGT was formulated with D230 in the same conditions, mixed in a 1:1 equivalent ratio. The mixture was cured at 100 °C for 2 h, 130 °C for 2 h and 150 °C for 2 h to achieve fully cured polymers. All the samples were cured under the same conditions and carefully removed from the mold for properties study.
2.6 Characterization
Fourier transform infrared (FT-IR) spectra were recorded on a Thermo Nicolet 6700 Fourier transform infrared spectrometer from Thermo-Fisher Scientific, scanning from 400 to 4000 cm−1 and 32 scans were collected for each sample. NMR spectrum was recorded on a Bruker 400 AVANCE III spectrometer operating at 400 MHz. It was conducted at 25 °C with the solvent of CDCl3 or acetone-d6.The DSC measurement was conducted on a METTLER TOLEDO-TGA/DSC I under a nitrogen atmosphere. Approximately 5–10 mg of each sample (epoxy monomer mixed with curing agent) was weighed and sealed in 40 μL aluminum crucibles. The samples were heated from 25 °C to 200 °C at heating rates of 5, 10, 15 and 20 °C min−1, respectively. The curves of heating flow as a function of time were recorded for activation energy analysis. DSC analysis for each sample was repeated twice. TGA measurement was conducted on a Mettler-Toledo TGA/DSC1 thermogravimetric analyzer (METTLER TOLEDO, Switzerland) with high purity nitrogen as purge gas at a scanning rate of 20 °C min−1 from 50 to 800 °C. The sample weighted about 5 mg was applied for each test. Degradation temperatures at 5% (Td5) and 30% (Td30) weight loss and the char yield at 800 °C (R800) were recorded for the cured resins. DMA was performed on Mettler-Toledo DMA/SDTA861e under a three points bending mode at a frequency of 1 Hz. The test samples were scanned from 25 °C to 200 °C at a heating rate of 3 °C min−1 and the glass transition temperature (Tg) was defined as the peak temperature of tanδ curves. For each sample, multiple tests were performed in order to ensure the accuracy.
The mechanical properties of the cured resins were carried out on an Instron 5567 Electric Universal Testing Machine (Instron, America). The tensile properties of the specimens with the gauge length of 50 mm were measured at a speed of 10 mm min−1 according to GB/T 1040.2-2006. The flexural strength was evaluated by means of a three-point bending test at a speed of 2 mm min−1 and five replicates were tested for each sample to obtain an average value.
3 Results and discussion
3.1 Chemical characterization of DGT and DGF
It is well known that there are two alternative procedures for the synthesis of epoxy resins. For the direct di-glycidylation, it is difficult for us to control the reaction accurately so as to obtain the products with specific chemical structures. Usually, the complicated oligomers with different degree of polymerization and epoxy values will be obtained and it is almost impossible to make the purification or separation. In this study, in order to compare the properties of epoxies derived from bio-based FDCA (DGF) and petroleum based terephthalic acid (DGT), the two step procedure was selected and it was shown in Scheme 3.The chemical structures of bis(prop-2-enyl)furan-2,5-dicarboxylate (FDCE), DGF and DGT were determined by means of FT-IR, 1H NMR and 13C NMR spectroscopy. The typical FT-IR spectrum of FDCE, DGF and DGT were presented in Fig. 1.
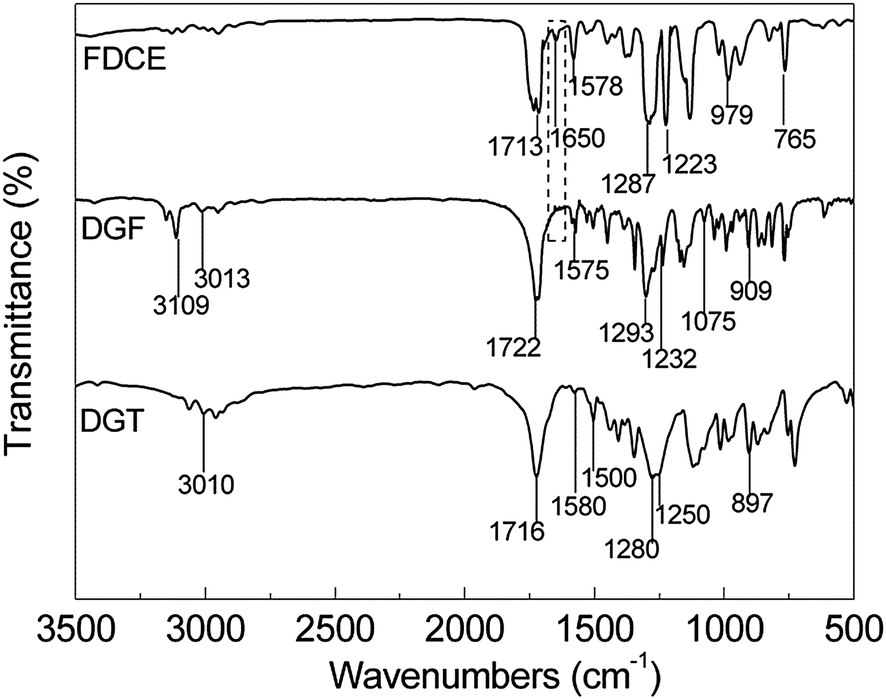 | ||
| Fig. 1 The FT-IR spectrum of FDCE, DGF and DGT. | ||
For the spectrum of FDCE, including the characteristic absorption bonds of furan rings (1578, 1223, 1018, 979, 825, 765 cm−1)53 the peaks arising from CO stretching vibration and C–O stretching were appeared at 1713 cm−1 and 1287 cm−1, respectively. No significant absorption in the –COOH stretching region was detected, which confirmed a high reaction yield between FDCA and allyl bromide. After FDCE was oxidized by m-CPBA, the characteristic peaks standing for the terminal CC bonds and CCH2 stretching absorption shown at 1650, 979 and 931 cm−1 were all disappeared. At the same time, the appearance of oxirane ring was indicated clearly by the arising peaks at 3013, 1232, 866 and 909 cm−1, reflecting the stretching of –CH2–, the stretching of C–O–C in oxirane group and the breathing absorption peak in oxirane group. The characteristic absorption bonds for the presence of oxirane ring and benzene ring of DGT were also detected in Fig. 1. Based on the FT-IR analysis, the structures of DGF and DGT could be confirmed preliminary. In order to make a further structure confirmation, the NMR spectrum of FDCE, DGF and DGT were shown in Fig. 2 and 3.
 | ||
| Fig. 2 1H NMR of FDCE (a), DGF (b) and DGT (c). | ||
 | ||
| Fig. 3 13C NMR spectra of FDCE (a), DGF (b) and DGT (c). | ||
In Fig. 2(a), the peaks between 4.5 and 6.5 ppm indicated that the allyl groups were introduced into FDCA with high purity and peak at 7.37 ppm was attributed to the furan ring's protons. In Fig. 2(b), the peaks at 2.73, 2.90 and 3.34 ppm represented the protons He, Hd and Hc in the ethylene oxide ring. The characteristic peaks for protons Ha and Hb were appeared at 4.67, 4.64, 4.20, 4.17 ppm, respectively. This complicated chemical shifts pattern for the protons is due to the presence of chiral carbon in the oxirane ring.37 In Fig. 2(c), the characteristic peaks for the protons in DGT were also assigned and the chemical shifts data were very much in line with the theoretical ones.
Fig. 3 is the 13C NMR spectra of FDCE, DGF and DGT. In Fig. 3(a), peaks at 132.1, 118.1 and 65.5 ppm represented the appearance of C3, C5 and C6 in the allyl group. The spectrum of DGF showed the disappearance of double bonds and the appearance of carbon atoms on the glycidyl epoxides at 65.9, 49.0 and 44.8 ppm in Fig. 3(b). This also indicated the oxidization of double bonds into oxirane rings. In Fig. 3(c), the spectrum of DGT displayed similar information to that of DGF. Therefore, it can be concluded that the result of the synthesis was indeed as expected.
3.2 Curing behavior and curing activity
Fig. 4 is the DSC thermograms of DGT/MHHPA, DGF/MHHPA, DGT/D230 and DGF/D230 systems at a heating rate of 5 °C min−1. All the systems displayed a single exothermic peak which was attributed to the ring opening reaction of epoxy groups in DGF and DGT with amino groups in D230 and anhydride group in MHHPA. The peak temperatures of curing curves might be taken as an indicator for the curing reactivity comparison of epoxy resin and curing agent.54 The higher the temperature of the peak is, the lower the reactivity is. Obviously, the peak temperatures of DGF systems were lower than those of DGT systems under the same curing conditions, no matter what kind of the curing agent was applied, MHHPA or D230. This result indicated that the reactivity of DGF was higher than that of DGT. | ||
| Fig. 4 DSC thermograms of DGT/MHHPA, DGF/MHHPA, DGT/D230 and DGF/D230 systems at a heating rate of 5 °C min−1. | ||
The curing reactivity of epoxy was greatly dependent on the heating rate and the shift of curing temperature with heating rate was a typical methodological phenomenon for non-isothermal curing. Therefore, the curing behaviors of DGF/DGT cured with MHHPA at different heating rates were investigated and both Kissinger's55 and Ozawa's56 methods were employed to calculate their activation energy (Ea) for accurate results. Fig. 5 shows the DSC curves of DGF and DGT cured with MHHPA at different heating rates. All the systems displayed single exothermic peaks with the different heating rates and the peak temperatures were increased with the accelerated heating rates.
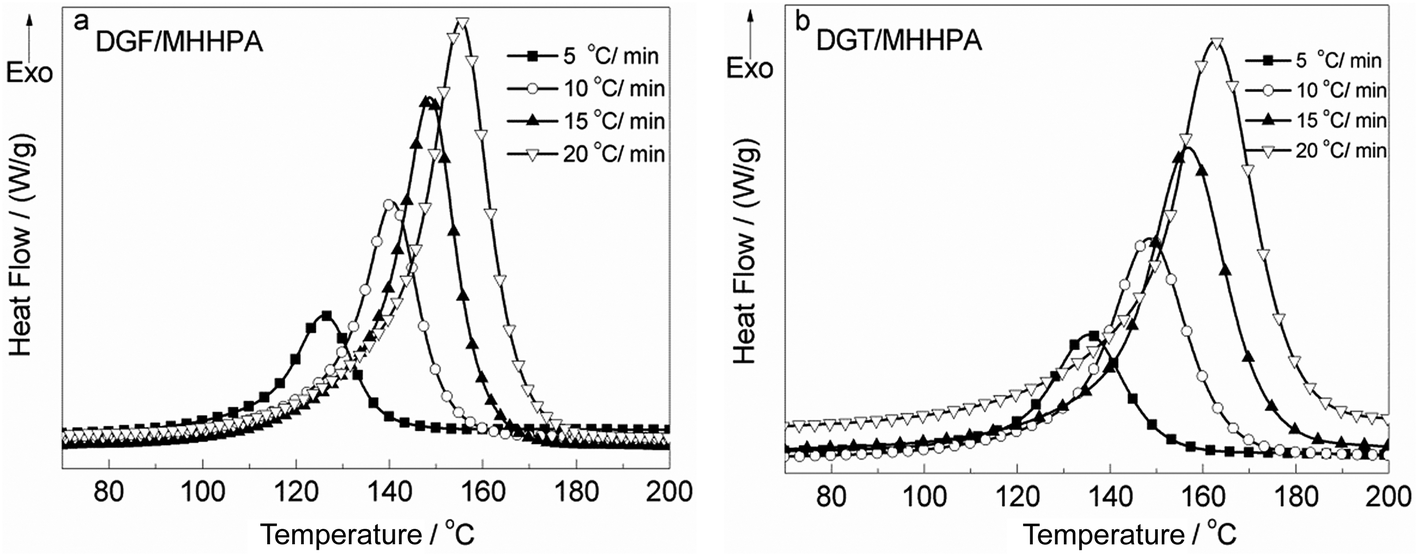 | ||
| Fig. 5 Typical DSC thermograms of DGF/MHHPA (a) and DGT/MHHPA (b) at different heating rate (5 °C min−1, 10 °C min−1, 15 °C min−1, 20 °C min−1). | ||
Curing activation energy was calculated based on the Kissinger's eqn (1):
| −ln(q/Tp2) = Ea/RTp − ln(AR/Ea) | (1) |
|
lnq = −1.052 × Ea/RTp + ln(AEa/R) − lnF(x) − 5.331
| (2) |
q versus 1/Tp.
Fig. 6 shows the plots of −ln(q/Tp2) versus 1/Tp based on Kissinger's equation (a) as well as lnq versus 1/Tp based on Ozawa's theory (b) for the DGT/MHHPA and DGF/MHHPA systems. Table 1 summarized the results and showed that DGF exhibited a little lower activation energy values compared with DGT, which indicated DGF has a higher curing reactivity.
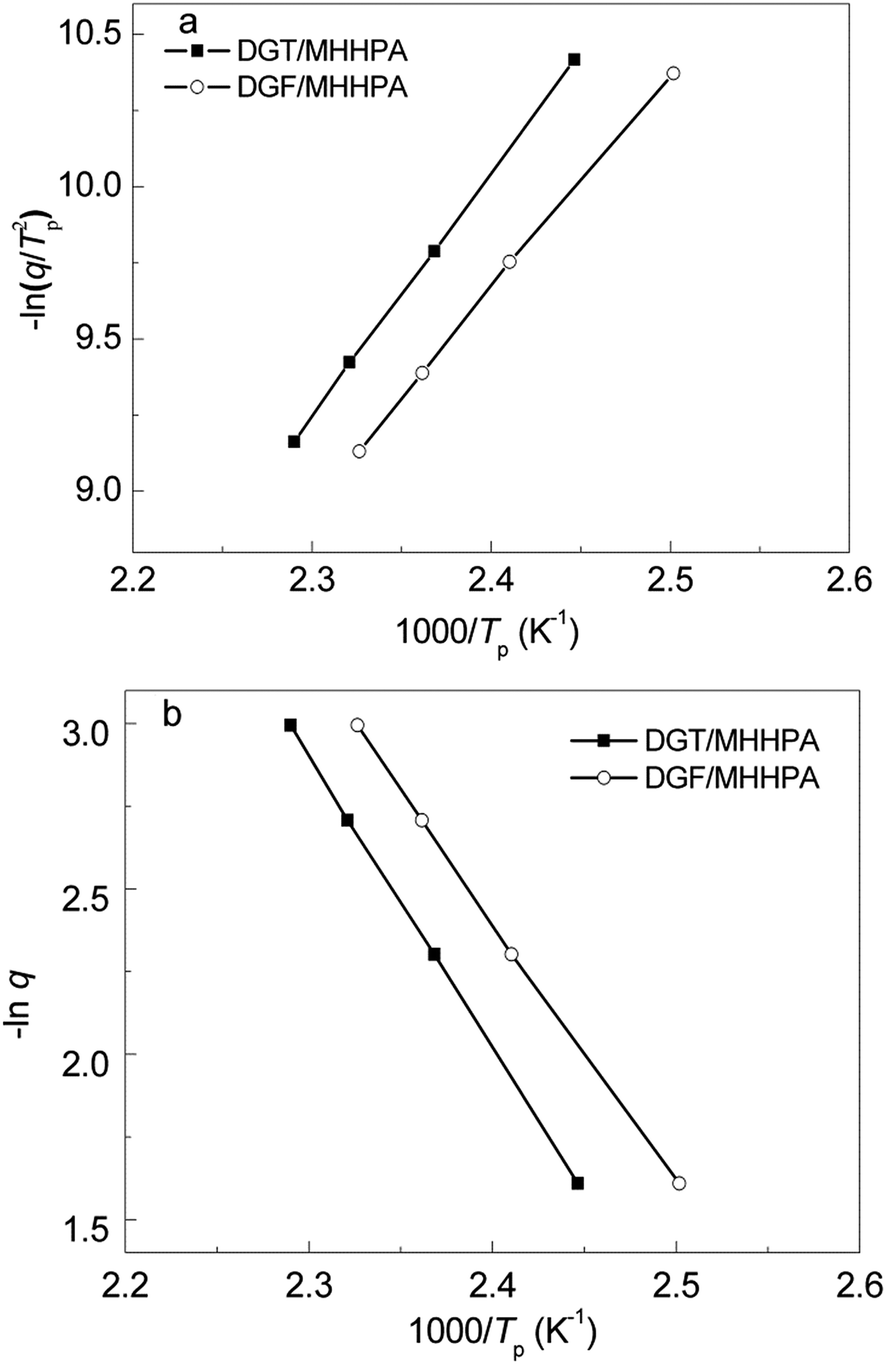 | ||
| Fig. 6 Linear plot of −ln(q/Tp2) versus 1/Tp based on Kissinger's equation (a) as well as lnq versus 1/Tp based on Ozawa's theory (b). | ||
| Samples | Heating rates (°C min−1) | Peak tem. (°C) | Ea (kJ mol−1) | |
|---|---|---|---|---|
| Kissinger | Ozawa | |||
| DGF/MHHPA | 5 | 126.6 | 58.82 | 62.43 |
| 10 | 141.7 | |||
| 15 | 150.3 | |||
| 20 | 156.7 | |||
| DGT/MHHPA | 5 | 135.6 | 66.39 | 69.78 |
| 10 | 149.1 | |||
| 15 | 157.7 | |||
| 20 | 163.5 | |||
The synthesized DGF and DGT were cured by the anhydride-based curing agent (MHHPA) and amine-based curing agent (D230), respectively. And the commonly used curing procedures (120 °C for 2 h, 150 °C for 2 h plus 180 °C for 2 h for DGF/MHHPA and DGT/MHHPA systems; 100 °C for 2 h, 130 °C for 2 h plus 150 °C for 2 h for DGF/D230 and DGT/D230 systems) for the similar systems were employed. In order to make sure that the samples were cured fully under this condition, the cured resins were applied to the DSC measurement (Fig. 7). Obviously, there were no exothermic peaks detected in the scanning curves at higher temperature, which verified the full curing process.
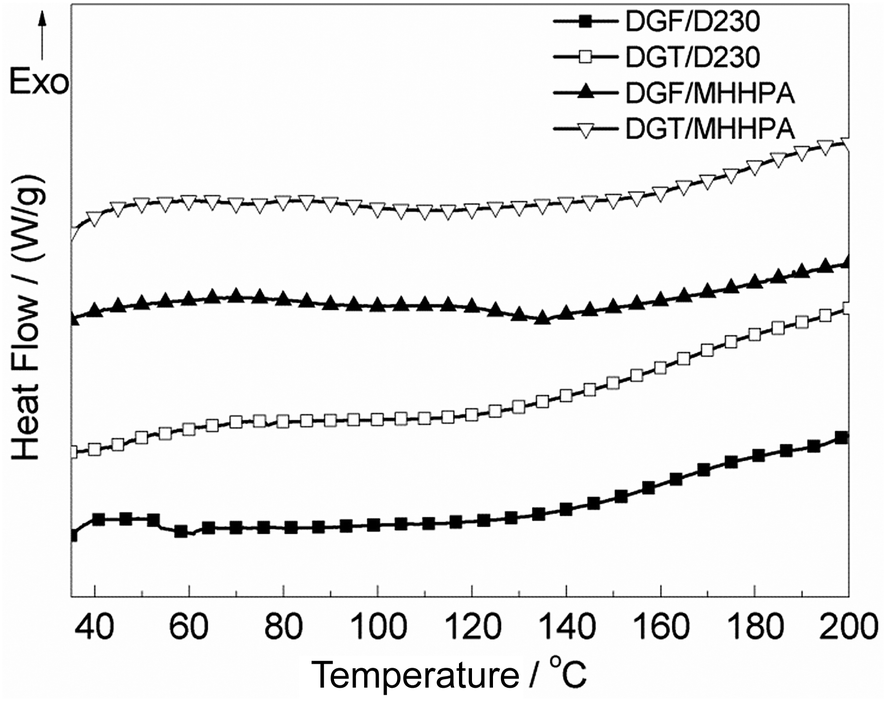 | ||
| Fig. 7 Heating DSC curves for the different cured resins. | ||
3.3 Dynamic mechanical properties of the epoxy networks
The dynamic mechanical analysis (DMA) was used to determine the glass transition temperature of the cured systems. Fig. 8 is the DMA plots of DGF and DGT cured with MHHPA and D230. As shown in Fig. 8(a), at a room temperature, DGF cured with MHHPA or D230 exhibited higher storage modulus when compared with that of DGT cured with the same curing agent. It is reported several times that the use of furan building block usually increases the stiffness of the resins.9,57,58 The relatively higher storage modulus of cured DGF systems might be due to the presence of furan ring structure in DGF systems. In Fig. 8(b), a sharp loss factor (tanδ) peak was noted in all cases and Tg is determined by the peak temperature of the tanδ curve. Apparently, the same trend was also notable where the DGF systems demonstrated significantly higher Tg than those of the DGT systems when cured with the same agents. The Tg data for the different samples were collected in Table 2. The glass transition temperature of epoxy networks is mainly dependent on its crosslink density and the chemical structure of the chain segment. The following equation for rubbery elasticity was employed here to calculate the crosslink density (ve) of the different cured systems:| E′ = 3veRT | (3) |
 | ||
| Fig. 8 DMA curves of storage modulus versus temperature (a) and tanδ versus temperature (b) for DGF/MHHPA, DGF/D230, DGT/MHHPA and DGT/D230. | ||
| Sample | DMA | Tensile (MPa) | Flexural (MPa) | ||||
|---|---|---|---|---|---|---|---|
| Tg (°C) | E′(MPa) Tg + 30 °C | ve (mol m−3) | Strength | Modulus | Strength | Modulus | |
| a Data for the DGEBA/MHHPA and DGEBA/D230 was obtained from the ref. 52 and 59. | |||||||
| DGF/MHHPA | 152 | 22 | 1930 | 84 ± 4 | 3000 ± 50 | 96 ± 3 | 3100 ± 110 |
| DGT/MHHPA | 128.8 | 18 | 1690 | 78 ± 2 | 3080 ± 80 | 90 ± 5 | 2950 ± 40 |
| DGF/D230 | 101.2 | 9 | 893 | 68 ± 3 | 2700 ± 110 | 75 ± 2 | 2500 ± 90 |
| DGT/D230 | 91.8 | 6 | 751 | 64 ± 2 | 2800 ± 60 | 73 ± 3 | 2400 ± 100 |
| DGEBA/MHHPA | 125 | NA | NA | 68 | 2900 | 135 | 3400 |
| DGEBA/D230 | 97 | NA | NA | NA | NA | 121 | 2950 |
3.4 Mechanical performance
The chemical structures of epoxy monomers and curing agents have significant influences on morphology and crosslink density of the cured networks, affecting their mechanical properties. The mechanical properties of cured DGF and DGT systems were tested by the Instron universal testing machine and the data were listed in Table 2. During the tests, all of the samples demonstrated the typical brittle fractures similar to those of the common thermosets. For the DGF/MHHPA system, the tensile strength, tensile modulus, flexural strength and flexural modulus (84 MPa, 3000 MPa, 96 MPa and 3100 MPa) were all similar or comparable to those of the DGT/MHHPA system (78 MPa, 3080 MPa, 90 MPa and 2950 MPa). When the more flexible curing agent D230 was applied, the predictable lower mechanical properties were shown and no significant difference was detected between DGF/D230 and DGT/D230 system. Besides compared with those of DGT, the mechanical properties of DGF/MHHPA and DGF/D230 have also been contrasted with the common diglycidyl ether of bisphenol A (DGEBA with the epoxide equivalent weight of 182–192 g eq.−1) cured with the same curing agents. As shown in Table 2, the FDCA based systems possessed the similar or comparable thermal and mechanical properties to those of the DGEBA based systems. According to above mechanical performance investigation and comparison, FDCA demonstrated a big chance to serve as the TPA substitution in the synthesis of epoxy resins with satisfied properties.3.5 Thermal properties
The thermal stability of cured epoxies in nitrogen was investigated by TGA and the typical curves for their thermal degradation behaviors were shown in Fig. 9. The values of the degradation temperature for 5% weight loss (Td5) and 30% weight loss (Td30) as well as the residual weight percent at 800 °C (R800) are presented in Table 3. In order to make a quantitative comparison of thermal stability, the statistic heat-resistant index (Ts) determined by the following equation was applied (4):60,61| Ts = 0.49[Td5 + 0.6(Td30 − Td5)] | (4) |
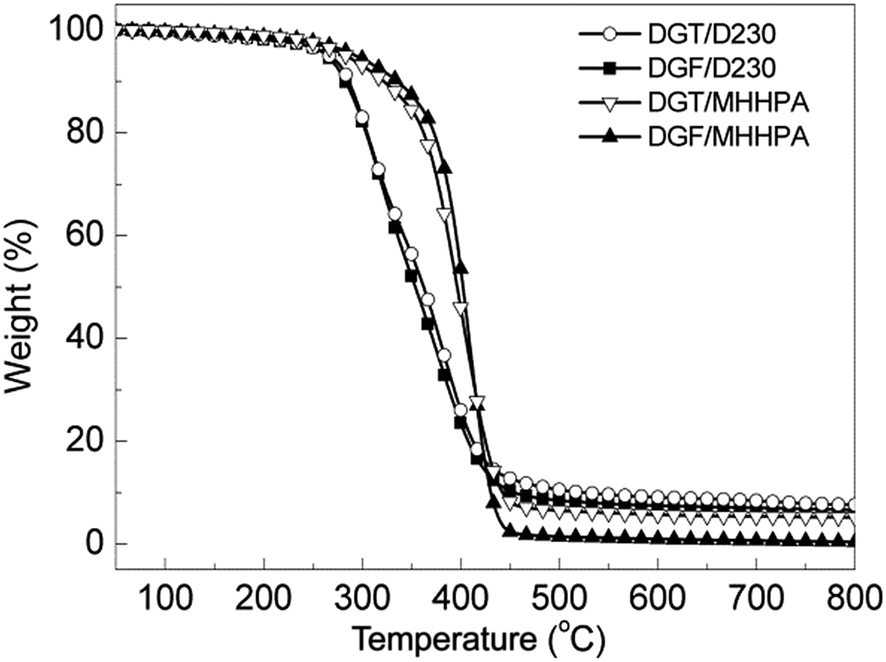 | ||
| Fig. 9 TGA curves of cured systems under N2 with a heating rate of 20 °C min−1. | ||
| Sample | Td5 (°C) | Td30 (°C) | R800 (%) | Ts |
|---|---|---|---|---|
| DGF/D230 | 267 | 320 | 6.8 | 146 |
| DGT/D230 | 266 | 322 | 8.2 | 147 |
| DGF/MHHPA | 293 | 381 | 1.9 | 169 |
| DGT/MHHPA | 284 | 378 | 5.2 | 167 |
4 Conclusions
A bio-based epoxy monomer DGF derived from 2,5-furandicarboxylic acid was synthesized by a two-steps method for the first time. For comparison, its petroleum-based analogue, diglycidyl ester of terephthalic acid (DGT) was also prepared. They were cured by the rigid (MHHPA) and soft (D230) curing agents respectively. Results showed that the DGF systems demonstrated higher curing reactivity, higher Tg, similar or comparable mechanical properties and thermal stability compared with those of DGT systems cured by the same curing agent. Bio-based FDCA showed great potential to replace the petroleum-based TPA in the synthesis of high performance epoxy. In the near future, FDCA might be an ideal renewable platform for the synthesis of not only thermoplastics but also thermosets.Acknowledgements
The authors greatly thank the financial support from the National Natural Science Foundation of China (NSFC no. 51373194), CAS-STS Project (no. KFJ-EW-STS-077).References
- A. Gandini, Macromolecules, 2008, 41, 9491–9504 CrossRef CAS.
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. J. Coelho, C. S. R. Freire and A. J. D. Silvestre, Polym. Chem., 2014, 5, 3119–3141 RSC.
- K. M. Nampoothiri, N. R. Nair and R. P. John, Bioresour. Technol., 2010, 101, 8493–8501 CrossRef PubMed.
- H. M. Muller and D. Seebach, Angew. Chem., Int. Ed. Engl., 1993, 32, 477–502 CrossRef.
- G. Impallomeni, A. Ballistreri, G. M. Carnemolla, S. P. P. Guglielmino, M. S. Nicolo and M. G. Cambria, Int. J. Biol. Macromol., 2011, 48, 137–145 CrossRef CAS PubMed.
- N. Jacquel, F. Freyermouth, F. Fenouillot, A. Rousseau, J. P. Pascault, P. Fuertes and R. Saint-Loup, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5301–5312 CrossRef CAS.
- R. M. Deshpande, V. V. Buwa, C. V. Rode, R. V. Chaudhari and P. L. Mills, Catal. Commun., 2002, 3, 269–274 CrossRef CAS.
- S. Chatti, S. M. Weidner, A. Fildier and H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2464–2471 CrossRef CAS.
- F. Hu, J. J. La Scala, J. M. Sadler and G. R. Palmese, Macromolecules, 2014, 47, 3332–3342 CrossRef CAS.
- J. Lewkowski, Arkivoc, 2001, 1, 17–54 Search PubMed.
- A. Gandini, Polym. Chem., 2010, 1, 245–251 RSC.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass. Volume I—Results of Screening for Potential Candidates from Sugars and Synthesis Gas, N. R. E. Laboratory Report DOE/GO-102004-1992, U.S. Department of Energy, 2004 Search PubMed.
- J. A. Moore and J. E. Kelly, Macromolecules, 1978, 11, 568–573 CrossRef CAS.
- M. Jiang, Q. Liu, Q. Zhang, C. Ye and G. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1026–1036 CrossRef CAS.
- E. Gubbels, L. Jasinska-Walc and C. E. Koning, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 890–898 CrossRef CAS.
- R. Storbeck and M. Ballauff, Polymer, 1993, 34, 5003–5006 CrossRef CAS.
- A. Gandini, A. J. D. Silvestre, C. P. Neto, A. F. Sousa and M. Gomes, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 295–298 CrossRef CAS.
- A. Mitiakoudis and A. Gandini, Macromolecules, 1991, 24, 830–835 CrossRef CAS.
- A. Gandini and M. N. Belgacem, Prog. Polym. Sci., 1997, 22, 1203–1379 CrossRef CAS.
- C. Moreau, M. Belgacem and A. Gandini, Top. Catal., 2004, 27, 11–30 CrossRef CAS.
- E. K. Moss, J. Cell. Plast., 1982, 18, 240–244 CrossRef CAS PubMed.
- S. Boufi, M. N. Belgacem, J. Quillerou and A. Gandini, Macromolecules, 1993, 26, 6706–6717 CrossRef CAS.
- R. A. Azzam, S. K. Mohamed, R. Tol, V. Everaert, H. Reynaers and B. Goderis, Polym. Degrad. Stabil., 2007, 92, 1316–1325 CrossRef CAS PubMed.
- P. Du, M. Wu, X. Liu, Z. Zheng, X. Wang, P. Sun, T. Joncheray and Y. Zhang, New J. Chem., 2014, 38, 770–776 RSC.
- E. DeJong, R. Dam, L. Sipos, D. Den Ouden and G. J. Gruter, 241st National Meeting and Exposition of the American-Chemical-Society, ACS, North Holland, Netherlands, 2011 Search PubMed.
- S. Flint, T. Markle, S. Thompson and E. Wallace, J. Environ. Manage., 2012, 104, 19–34 CrossRef CAS PubMed.
- R. Wool and X. S. Sun, in Bio-Based Polymers and Composites, ed. R. Wool and X. S. Sun, Academic Press, Burlington, 2005 Search PubMed.
- L. M. Bonnaillie and R. P. Wool, J. Appl. Polym. Sci., 2007, 105, 1042–1052 CrossRef CAS.
- D. P. Pfister and R. C. Larock, Bioresour. Technol., 2010, 101, 6200–6206 CrossRef CAS PubMed.
- Y. S. Lu and R. C. Larock, J. Appl. Polym. Sci., 2006, 102, 3345–3353 CrossRef CAS.
- V. Sharma and P. P. Kundu, Prog. Polym. Sci., 2006, 31, 983–1008 CrossRef CAS PubMed.
- K. Huang, P. Zhang, J. Zhang, S. Li, M. Li, J. Xia and Y. Zhou, Green Chem., 2013, 15, 2466 RSC.
- G. Lligadas, J. C. Ronda, M. Galia and V. Cadiz, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5630–5644 CrossRef CAS.
- K. Hofmann and W. Glasser, Macromol. Chem. Phys., 1994, 195, 65–80 CrossRef CAS.
- C. Sasaki, M. Wanaka, H. Takagi, S. Tamura, C. Asada and Y. Nakamura, Ind. Crops Prod., 2013, 43, 757–761 CrossRef CAS PubMed.
- X. Q. Liu, W. B. Xin and J. W. Zhang, Green Chem., 2009, 11, 1018–1025 RSC.
- X. Q. Liu and J. W. Zhang, Polym. Int., 2010, 59, 607–609 CrossRef CAS.
- C. Li, X. Liu, J. Zhu, C. Zhang and J. Guo, J. Macromol. Sci., Part A: Pure Appl.Chem., 2013, 50, 321–329 CrossRef CAS.
- L. Deng, C. Ha, C. Sun, B. Zhou, J. Yu, M. Shen and J. Mo, Ind. Eng. Chem. Res., 2013, 52, 13233–13240 CrossRef CAS.
- P.-H. Elchinger, D. Montplaisir and R. Zerrouki, Carbohydr. Polym., 2012, 87, 1886–1890 CrossRef CAS PubMed.
- N. D. Sachinvala, D. L. Winsor, R. K. Menescal, I. Ganjian, W. P. Niemczura and M. H. Litt, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 2397–2413 CrossRef CAS.
- L. J. Cao, X. Q. Liu, H. N. Na, Y. G. Wu, W. G. Zheng and J. Zhu, J. Mater. Chem. A, 2013, 1, 5081–5088 CAS.
- C. Aouf, H. Nouailhas, M. Fache, S. Caillol, B. Boutevin and H. Fulcrand, Eur. Polym. J., 2013, 49, 1185–1195 CrossRef CAS PubMed.
- R. X. Chang, J. L. Qin and J. G. Gao, J. Polym. Res., 2014, 21, 501 CrossRef.
- A. Gandini, Green Chem., 2011, 13, 1061–1083 RSC.
- R. Auvergne, S. Caillol, G. David, B. Boutevin and J.-P. Pascault, Chem. Rev., 2013, 114, 1082–1115 CrossRef PubMed.
- J. N. Xin, P. Zhang, K. Huang and J. W. Zhang, RSC Adv., 2014, 4, 8525–8532 RSC.
- G. Mashouf Roudsari, A. K. Mohanty and M. Misra, ACS Sustainable Chem. Eng., 2014, 2, 2111–2116 CrossRef CAS.
- P. Y. Kuo, M. Sain and N. Yan, Green Chem., 2014, 16, 3483–3493 RSC.
- M. Stemmelen, F. Pessel, V. Lapinte, S. Caillol, J. P. Habas and J. J. Robin, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2434–2444 CrossRef CAS.
- J. L. Qin, H. Z. Liu, P. Zhang, M. Wolcott and J. W. Zhang, Polym. Int., 2014, 63, 760–765 CrossRef CAS.
- S. Q. Ma, X. Q. Liu, Y. H. Jiang, Z. B. Tang, C. Z. Zhang and J. Zhu, Green Chem., 2013, 15, 245–254 RSC.
- Q. Tian, M. Z. Rong, M. Q. Zhang and Y. C. Yuan, Polym. Int., 2010, 59, 1339–1345 CrossRef CAS.
- W. B. Liu, Q. H. Qiu, J. Wang, Z. C. Huo and H. Sun, Polymer, 2008, 49, 4399–4405 CrossRef CAS PubMed.
- H. E. Kissinger, J. Res. Natl. Bur. Stand., 1956, 57, 217–221 CrossRef CAS.
- T. Ozawa, J. Therm. Anal., 1976, 9, 369–373 CrossRef CAS.
- A. F. Sousa, M. Matos, C. S. R. Freire, A. J. D. Silvestre and J. F. J. Coelho, Polymer, 2013, 54, 513–519 CrossRef CAS PubMed.
- P. Pan, W. Kai, B. Zhu, T. Dong and Y. Inoue, Macromolecules, 2007, 40, 6898–6905 CrossRef CAS.
- S. Q. Ma, X. Q. Liu, L. B. Fan, Y. H. Jiang, L. J. Cao, Z. B. Tang and J. Zhu, ChemSusChem, 2014, 7, 555–562 CrossRef CAS PubMed.
- R. S. Lehrle and R. J. Williams, Macromolecules, 1994, 27, 3782–3789 CrossRef CAS.
- E. Khosravi and O. M. Musa, Eur. Polym. J., 2011, 47, 465–473 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |