
Synthesis of Ag/ZnO/C plasmonic photocatalyst with enhanced adsorption capacity and photocatalytic activity to antibiotics
Abstract
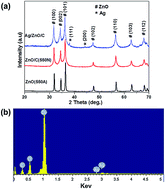
A novel Ag/ZnO/C plasmonic photocatalyst was synthesized via a facile calcination and photodeposition route. Samples were characterized by X-ray diffraction (XRD), energy dispersive X-ray spectroscopy (EDS), transmission electron microscopy (TEM) and ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS). The results indicated that Ag and ZnO nanoparticles sized 5–10 nm were uniformly dispersed on the surface of the carbonaceous layers in Ag/ZnO/C composites. The adsorption capacity and photocatalytic activity were investigated by adsorption and photocatalytic degradation of tetracycline hydrochloride (TC-HCl) in aqueous solution. The results showed that the obtained Ag/ZnO/C sample exhibited higher adsorption capacity and enhanced UV and visible light driven photocatalytic activity to TC-HCl compared to ZnO/C and pure ZnO. With the presence of Ag nanoparticles and carbonaceous layers incorporated in the structure, the Ag/ZnO/C composites can make use of not only the UV region of sunlight, but also the visible region and efficiently promote photogenerated electron separation and transportation as well as generating more active reaction sites, which synergistically facilitate the photocatalysis process. Our present work provides a simple and new pathway for the design of ZnO-based catalysts that respond to both UV and visible light and promotes their practical application in various environmental and energy issues driven by solar light.
 Please wait while we load your content...
Please wait while we load your content...

