
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
BF3-promoted electrochemical properties of quinoxaline in propylene carbonate†
Emily V.
Carino
ab,
Charles E.
Diesendruck‡
ac,
Jeffrey S.
Moore
ac,
Larry A.
Curtiss
ad,
Rajeev S.
Assary
*ad and
Fikile R.
Brushett
*ab
aJoint Center for Energy Storage Research, USA. E-mail: assary@anl.gov; brushett@mit.edu; Tel: +1-630-252-3536 Tel: +1-617-324-7400
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
cDepartment of Chemistry, University of Illinois at Urbana Champaign, Urbana, IL 61801, USA
dMaterials Science Division, Argonne National Laboratory, Argonne, IL 60439, USA
First published on 4th February 2015
Abstract
Electrochemical and density functional studies demonstrate that coordination of electrolyte constituents to quinoxalines modulates their electrochemical properties. Quinoxalines are shown to be electrochemically inactive in most electrolytes in propylene carbonate, yet the predicted reduction potential is shown to match computational estimates in acetonitrile. We find that in the presence of LiBF4 and trace water, an adduct is formed between quinoxaline and the Lewis acid BF3, which then displays electrochemical activity at 1–1.5 V higher than prior observations of quinoxaline electrochemistry in non-aqueous media. Direct synthesis and testing of a bis-BF3 quinoxaline complex further validates the assignment of the electrochemically active species, presenting up to a ∼26-fold improvement in charging capacity, demonstrating the advantages of this adduct over unmodified quinoxaline in LiBF4-based electrolyte. The use of Lewis acids to effectively “turn on” the electrochemical activity of organic molecules may lead to the development of new active material classes for energy storage applications.
1. Introduction
Stationary energy storage systems are needed to facilitate the widespread penetration of intermittent renewable electricity generators such as solar photovoltaic and wind turbines, and to improve energy efficiency of the electric grid.1 Redox flow batteries (RFBs) may offer the best combination of cost, performance, and operational flexibility to meet these needs.2 Unlike enclosed rechargeable batteries which house all components in a single cell, RFBs utilize the reduction and oxidation of electro-active species in flowable solutions or suspensions that are housed in external tanks and pumped to a power-converting electroreactor. To date, the vast majority of flow battery technologies are based on aqueous electrochemistry, with all-vanadium and zinc–bromine systems being the most successful.2d,3Redox-active organic molecules are particularly promising charge storage materials for RFBs because relevant properties like potential and solubility can be tuned through modifications of molecular structure. Aziz and co-workers recently described a bench-scale aqueous flow battery utilizing quinones as charge storage materials.4 Furthermore, they employed quantum chemical computations to show a correlation between reduction potential and the degree of substitution with hydroxyl groups and demonstrated good agreement with experimental measurements in sulfuric acid media.4 While the thermodynamic stability window of water is 1.23 V, sluggish hydrogen and oxygen evolution kinetics on electrode materials can enable higher cell voltages, for example lead-acid and zinc–bromine batteries. Deploying non-aqueous solvents offers an even wider window of electrochemical stability which enables non-aqueous RFBs to operate at cell potentials >3.0 V, thereby leading to higher energy density and typically higher roundtrip efficiency, which together reduce the cost of energy. To take full advantage of this extended potential window, electrochemical couples must be developed with suitably different reduction potentials, high solubility, and good long term stability. To date, a selection of non-aqueous electrochemically active materials have been studied including transition metal centered coordination complexes,5 transition metal centered ionic liquids,6 and organic molecules.7
Quinoxalines are a promising family of redox-active materials due to their high intrinsic capacity (ca. 410 mA h g−1 for quinoxaline, assuming 2e− transfer) and high solubility in carbonate solvents (∼7 M). Recently, Brushett et al. investigated a variety of quinoxaline derivatives as the negative electrolyte active species (anolyte) for non-aqueous Li-ion RFBs and observed two coupled electroreduction and oxidation events between 2.4–3.2 V vs. Li/Li+ in an electrolyte consisting of 0.2 M lithium tetrafluoroborate (LiBF4) in propylene carbonate.8 Interestingly, Ames et al. reported a single redox event between 1.4–2.0 V vs. Li/Li+ for similar derivatives in 0.1 M tetraethylammonium perchlorate (TEAP) in dimethylformamide.9 Comparable results to those of Ames et al. were obtained by Barqawi et al.10 in 0.1 M tetraethylammonium hexafluorophosphate (TEAPF6) in acetonitrile and Angulo et al.11 in 0.1 M tetra-n-butylammonium hexafluorophosphate (TBAPF6) in acetonitrile or dichloromethane.
To design and optimize quinoxalines for non-aqueous RFBs, their electronic properties as well as interactions with different electrolytes must be understood, controlled, and eventually manipulated. Moreover, given the sheer number of possible quinoxaline derivatives and electrolyte compositions, incorporating an understanding of electrolyte interactions into molecular design rules is critical to guiding future research directions. Here we describe the role of the Lewis acid BF3 in the electrochemical behavior of quinoxaline in LiBF4–propylene carbonate electrolyte solutions. We used electrochemical studies in combination with explicit computational models to demonstrate the effect of various electrolyte constituents on the voltammetry and cycling behavior of quinoxaline. First, we characterized the effect of electrolyte composition and decomposition products on the electrochemical behavior of quinoxaline. Second, we modeled the impact of electron-donating and electron-withdrawing substituent groups, as well as adducts formed from different types of electrolyte salts, on the thermodynamic and related electrochemical properties of quinoxalines. Third, the confluence of these experimental and computational efforts resulted in the design and synthesis of a novel substituted quinoxaline molecule exhibiting a 26-fold improvement in charging capacity compared to bare quinoxaline.
2. Experimental
2.1 Electrochemical details
Anhydrous propylene carbonate (PC, 99.7%, Sigma-Aldrich), acetonitrile (ACN, 99.8%, Sigma-Aldrich), quinoxaline (Q, 99.0%, Sigma-Aldrich), 2,3,6-trimethylquinoxaline (2,3,6-TMQ, 97.0%, Alfa Aesar), anhydrous lithium tetrafluoroborate (LiBF4, 98.0%, Acros Organics), lithium trifluoromethansulfonate (Li triflate, 99.5%, Sigma-Aldrich), lithium bis(perfluoroethylsufonyl)imide (Li BETI, 3 M), tetrabutylammonium tetrafluoroborate (TBABF4, 99.0%, Fluka), tetraethylammonium tetrafluoroborate (TEABF4, >99.0%, Fluka), lithium perchlorate (LiClO4, 99.99% trace metals basis, Aldrich), lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, 99.95% trace metals basis, Aldrich), sodium tetrafluoroborate (NaBF4, 97%, Sigma-Aldrich) and boron trifluoride diethyletherate (BF3·OEt2, 46.5% BF3, Sigma-Aldrich) were used as received. All chemicals except BF3·OEt2 were opened and stored in an argon (Ar)-filled glovebox (MBraun Labmaster). When not in use the BF3·OEt2 was stored in a refrigerator, and was used only inside of the glovebox. All electrolyte solutions were prepared in the glovebox. The water content of solutions for electrochemical measurements was quantified with Karl-Fischer titration by a C20 Coulometric K-F Titrator (Mettler-Toledo) with Hydranal® Coulomat-AG (Fluka) as the reagent.Electrochemical experiments were performed in the glovebox using either a CHI760E potentiostat (CH Instruments, Inc.) or a 1470E Solartron Analytical Instrument. Two different 3-electrode electrochemical cells were used in this report. An electrochemical cell consisting of a 3 mm diameter glassy carbon (GC) working electrode (CH Instruments, Inc.), a lithium metal counter electrode, and a lithium metal reference electrode was used for collecting the data displayed in Fig. 1. For all other cyclic voltammetry experiments, the electrochemical cell consisted of a 3 mm GC working electrode, a Pt wire counter electrode (CH Instruments, Inc.), and a Ag/Ag+ quasi-reference electrode (BASi) housed in a fritted glass container. Prior to use, the GC electrode was sequentially polished with 0.3 μm and 0.05 μm alumina grit on a MicroCloth pad (Buehler Ltd.), thoroughly rinsed with water that was deionized at 18.2 MΩ (Millipore) and dried under Ar before transfer to the glovebox. The reference electrode compartment, a Vycor™ fritted glass tube, was filled with a saturated solution of AgNO3 in PC (Sigma Aldrich). The reference electrode potential was determined using ferrocene as an internal standard. The internal standard was measured after each sample to account for drift in the reference electrode potential. All potentials for electrochemical experiments are reported versus the Li/Li+ reference couple (−3.04 V vs. SHE), and were placed on the Li/Li+ reference scale by directly measuring the formal potential of ferrocene (0.64 V vs. SHE, 3.68 V vs. Li/Li+). Constant-current chronopotentiometry was performed in a bulk electrolysis cell (BASi, Inc., Indiana) comprised of a reticulated vitreous carbon working electrode, a Li foil counter electrode held in a fritted compartment, and a Li foil reference electrode housed in a fritted glass tube (BASi, Inc., Indiana) filled with a 1 M LiClO4/PC electrolyte. For studies involving the addition of water to non-aqueous solutions, the reference electrode consisted of a Li metal foil housed in a Vycor-fritted tube (BASi) filled with a 1 M LiTFSI/PC electrolyte. This configuration was used to prevent water from attacking the reference electrode. Pt and Au working electrodes (2 mm diameter, CH Instruments) were used in some studies presented in the ESI.†
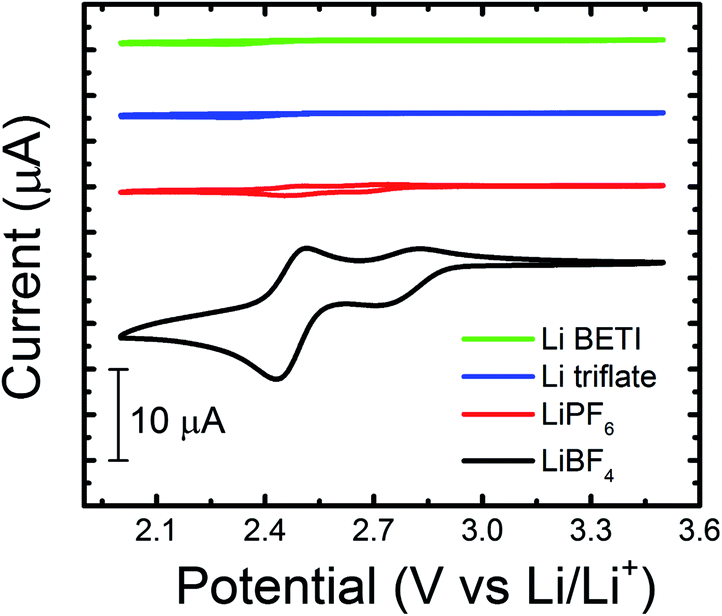 | ||
| Fig. 1 Cyclic voltammograms of 5 mM 2,3,6-TMQ in various 0.2 M Li-ion salt/PC electrolytes. All experiments were performed in a GC/Li/Li cell at a scan rate of 10 mV s−1. | ||
2.2 Computational details
We employed the B3LYP/6-31+G(d) level of theory to compute the structure and energetics of all species using Gaussian 09 Software. The same level of theory was used to calculate zero point energies, free energy corrections (298 K, 1 atm pressure) and solvation energies. The SMD model was used to compute the solvation free energy by a single point energy calculation on the gas phase optimized geometry using water as the dielectric medium.12 We find that this is an effective approximation for computing free energies of electrochemically active species in solution. We have optimized selected systems using the SMD solvent with a water dielectric medium model to include the solvation effects in determining the geometry and energy. For this study, changing the dielectric medium to acetone, dimethylsulfoxide, or methanol did not significantly affect the computed reduction potentials of quinoxaline derivatives (Table S1†). The Gibbs free energy (298 K) in solution is computed as the sum of the free energy in the gas phase and the solvation energy. Upon computing the solution phase free energy change for reduction or oxidation process (ΔGredox), the reduction potential (EVredox) is calculated via the following identity, EVredox = −ΔGredox/nF, where n is the number of electrons involved in the reduction reaction and F is the Faraday constant. Thereafter, the computed reduction potential is referenced to a Li/Li+ electrode, a typical standard used for non-aqueous Li-ion electrochemistry, via the following equation, EVredox (Li/Li+) = EVredox − 1.24 V, where 1.24 V represents the difference between the standard hydrogen electrode (SHE, −4.28 V (ref. 13)) and Li/Li+ reduction couple (−3.04 V). The addition of the constant ‘−1.24 V’ is required to convert the free energy changes to reduction potential (Li/Li+ reference electrode), a commonly used convention to compute the reduction potentials in solution.14 The change in electron energy when going from vacuum to non-aqueous solution is treated as zero, similar to other reports.15 Further details regarding the computation of reduction potential can be found elsewhere.15,16It should be noted that the binding of the second electron to the mono-anion in the gas phase is thermodynamically uphill (negative electron affinity), while inclusion of solvation contributions favors the binding of the second electron. The negative electron affinities result in less accurate reduction potential, but agreement is reasonable in cases where experimental values are available. It was found that finite basis sets gives reasonable results in comparison to gas phase experimental results for gas phase temporary anions with negative electron affinities due to a cancellation of errors.17 In general, quantum chemical calculations are able to compute the influence of different salt and solvent molecules on the reduction potential of a material of interest.16h,18
2.3 Synthesis of quinoxaline (bis)trifluoroborane
Unless otherwise stated, all chemical reagents were obtained from commercial suppliers and used without purification. Solvents were purified in a solvent purification system with alumina columns. The synthesis procedure was modified from Martin et al.19 Quinoxaline 0.5 g (3.8 mmol) was dissolved in 50 mL anhydrous dichloromethane (CH2Cl2, Sigma-Aldrich) in a Schlenk flask under dry nitrogen; the flask was then immersed in liquid nitrogen. To this, 3.16 mL (25.0 mmol) of BF3·OEt2 were injected by a syringe through a rubber septum. The etherate froze as a top layer in the flask. The contents of the flask were allowed to warm slowly to room temperature with stirring, during which a yellow solid precipitated. The reaction mixture was stirred for additional 3 h after which the flask was brought inside a glove-box where the solid was collected on a vacuum filter and washed with ca. 10 mL CH2Cl2. The solid was then dried under vacuum to give 538 mg (52.6% yield) of the complex as a yellow solid. 1H, 13C, 11B and 19F NMR spectra were collected using a Varian 500 or 400 MHz spectrometer in the VOICE NMR laboratory at the University of Illinois; the residual solvent proton and carbon, and BF3·OEt2 were used to reference the chemical shifts. High-resolution electrospray ionization (HR-ESI) mass spectra were obtained through the Mass Spectrometry Facility, School of Chemical Sciences, University of Illinois. For the yellow solid: 1H NMR (500 MHz, CD3CN): δ (ppm) 9.29 (s, 2H), 8.53 (dd, J = 3.2 Hz, 2H), 8.18 (dd, J = 3.2 Hz, 2H). 13C NMR (125 MHz, THF-d8): δ (ppm) 146.2, 145.9, 131.9, 130.0. 11B (128 MHz, CD3CN): δ (ppm) −1.6. 19F (470 MHz, CD3CN): δ (ppm) 145.7. HRMS-ESI (m/z): calculated for C8H7BF3N2 [M − BF3 + H]+: 199.0649; found: 199.0634.3. Results and discussions
3.1 Electrochemical analysis of quinoxaline–electrolyte interactions
Quinoxaline is electrochemically active at 1.4 V vs. Li/Li+ in ACN, in accordance with previous results and theoretical predictions (vide infra), yet does not display comparable electrochemical activity in PC (ESI, Fig. S1†). Our previous studies revealed that quinoxaline derivatives are electrochemically active in carbonate-based electrolytes at more positive potentials (ca. 2 to 3 V vs. Li/Li+).8 To better understand the relationship between local electronic structure and experimentally measured properties, the electrochemical behavior of 2,3,6-trimethylquinoxaline (2,3,6-TMQ), the best performing quinoxaline derivative from ref. 8, was analyzed in the presence of four Li salts with different anions. Fig. 1 shows cyclic voltammograms of 2,3,6-TMQ in the presence of 0.2 M LiBF4, LiPF6, Li triflate, and Li BETI, all in PC, and clearly indicates that the choice of anion has a pronounced effect on the observed 2,3,6-TMQ electrochemical behavior. The largest reduction and oxidation currents are observed for 2,3,6-TMQ in 0.2 M LiBF4/PC electrolyte which also show two electron transfer events, consistent with previous results.8 Similarly, two electron transfer events are observed in 0.2 M LiPF6/PC, albeit at significantly lower currents. The current responses observed in 0.2 M Li triflate/PC and 0.2 M Li BETI/PC are comparable to those of the baseline electrolytes indicating no electrochemical activity in the scanned region. Interestingly, 2,3,6-TMQ did not show any activity above background current levels in 0.2 M TBABF4/PC (ESI, Fig. S2†) which suggests that the presence of the BF4− is not solely responsible for the activation. Changing the working electrode from GC to Pt or Au did not significantly alter the voltammetry of 2,3,6-TMQ in 0.2 M LiBF4/PC (ESI, Fig. S3†).To further study the promotional effect of LiBF4 on the reduction current of quinoxalines, the concentrations of two quinoxaline species (2,3,6-TMQ and quinoxaline) and LiBF4 were varied relative to each other. Fig. 2 shows the change in peak current of the well-defined second reduction wave (EPC-ii) which corresponds to the reduction wave at ∼2.45 V vs. Li/Li+ (Fig. 1) of 2,3,6-TMQ and quinoxaline as a function of active species and LiBF4 concentration. Fig. 2a shows that increasing the 2,3,6-TMQ concentration from 5 to 50 mM, while holding the LiBF4 concentration constant at 0.2 M, results in only slightly more than a two-fold increase in the current. In comparison, increasing the LiBF4 concentration, while holding 2,3,6-TMQ concentration constant at 50 mM, leads to directly proportional increases in observed current. Similar trends were observed for quinoxaline (denoted as Q in Fig. 2b). The voltammetry corresponding to these data points is presented in the ESI (Fig. S4†). These results indicate that the magnitude of the reduction current is due to an interaction between the active species and supporting electrolyte which, at these concentrations, has a stronger dependence on salt concentration.
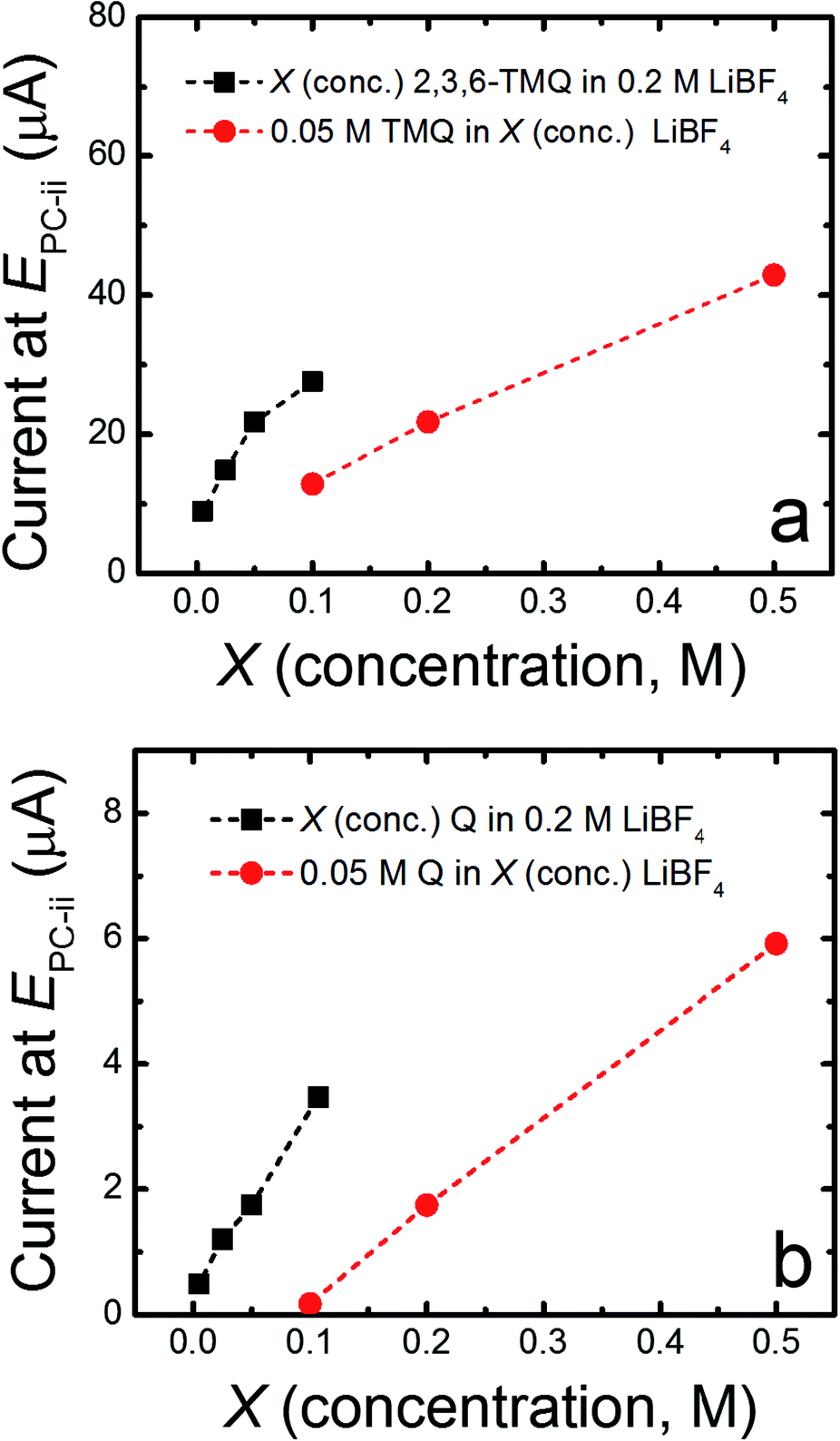 | ||
| Fig. 2 Peak reduction current of the 2nd wave of 2,3,6-TMQ (a) and quinoxaline, Q (b) as a function of either active species and LiBF4 concentration. In all experiments, PC was used as a solvent. Peak currents were obtained from CV measurements taken in a GC/Pt/Ag/Ag+ cell at a scan rate of 10 mV s−1. | ||
LiBF4 weakly dissociates in PC due to the strong ion-pairing between Li+ and BF4−.20 LiBF4 also decomposes into BF3via thermolysis and hydrolysis with trace water.21 BF3, a strong Lewis acid, is expected to complex with quinoxaline, which acts as a Lewis base. LiPF6, a common Li-ion battery salt, undergoes a similar thermal decomposition process (LiPF6 ↔ LiF + PF5) which in turn leads to a number of undesirable reactions that limit the battery life.22 Others have shown that introduction of Lewis base additives (e.g., pyridine) dramatically increases the thermal stability of the electrolyte by binding the reactive PF5 intermediates.23
To determine if the presence of BF3 is linked to the observed reduction current of quinoxalines, we spiked the electrolytes consisting of 2,3,6-TMQ or quinoxaline in Li triflate/PC with BF3·OEt2 (Fig. 3). Recall that 2,3,6-TMQ did not display any significant electrochemical behavior in the Li triflate/PC electrolyte (see Fig. 1). Fig. 3a shows the impact of adding 0.2 M BF3·OEt2 to electrolyte solutions consisting of 5 mM 2,3,6-TMQ in 0.5 M Li triflate/PC (red line) and 0.5 M LiBF4/PC (black line). In the presence of BF3 the observed reduction current is slightly increased and the two electron transfer waves are further separated and shifted towards more positive potentials. Fig. 3b shows the impact of adding 0.1 M BF3·OEt2 to electrolyte solutions consisting of 5 mM quinoxaline (denoted as Q in the figure) in 0.2 M Li triflate/PC (red line) and 0.2 M LiBF4/PC (black line). In this case, the addition of BF3·OEt2 has a more dramatic effect on the observed electrochemical behavior. Specifically, the reduction current increases by an order of magnitude and the voltammogram includes new reduction and oxidation peaks spaced closely together in energy. Moreover, the addition of BF3·OEt2 to solutions containing only quinoxaline in PC without any supporting electrolyte resulted in reduction currents well above the background currents measured in solutions of PC containing only BF3·OEt2 (ESI, Fig. S5†).
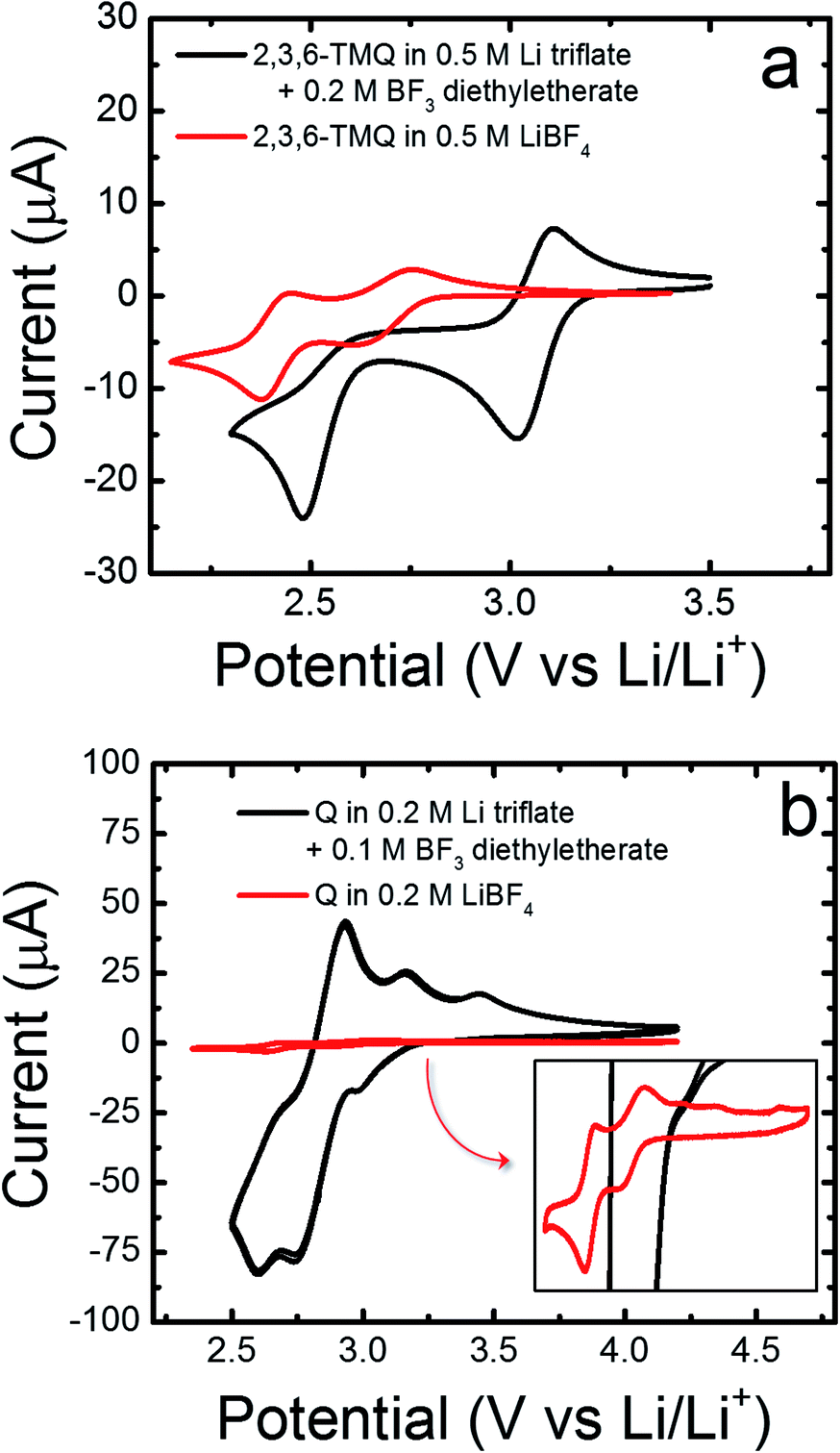 | ||
| Fig. 3 Effects of adding BF3, in the form of BF3·OEt2, on the redox activity of (a) 5 mM 2,3,6-TMQ, and (b) 5 mM quinoxaline (Q) at scan rates of 10 mV s−1 and 20 mV s−1, respectively. | ||
Quinoxaline is soluble and electrochemically active in select aqueous solutions;24 thus, it is imperative to consider the role of water contamination in the electrochemical behavior of quinoxalines in non-aqueous electrolytes. We note that the as-prepared (without subsequent drying) solutions contained significant amounts of water (>100 ppm), even when the solvent and electrolyte were dried using activated molecular sieves. Therefore, we dried some of the solutions of quinoxaline after mixing it with electrolyte and then compared the voltammetry to as-prepared solutions. Following drying with activated molecular sieves, the peak reduction current of 0.05 M quinoxaline in 0.5 M LiBF4 is only 0.5 μA (black line, Fig. S6†). This is approximately an order of magnitude lower than as-prepared solutions in which the water content is 150 ppm (red line, Fig. S6†). Although the voltammetry of the dried and as-prepared quinoxaline display similar features (two reduction and oxidation waves), the magnitude of the reduction current corresponding to the dried quinoxaline is comparable to that of the baseline current seen in the presence of TBABF4 (ESI, Fig. S2†), indicating that quinoxaline is barely electrochemically active in the absence of water. Importantly, the electrochemistry of quinoxaline in as-prepared Li triflate, which contained 200 ppm water, is comparable in magnitude to dried quinoxaline (Black line, Fig. S7†) and the background current as well (ESI, Fig. S2†), indicating that water alone does not render quinoxaline electrochemically active.
To better understand the role of water in the non-aqueous electrochemistry of quinoxaline, we examined the effects of intentional water contamination on quinoxaline voltammetry. Adding water to the dried quinoxaline/LiBF4 solution resulted in significant enhancement of the reduction current (ESI, Fig. S8a†) and lead to a similar voltammetric fingerprint as described earlier by Brushett and co-workers8 and is supported here by our computational results. Adding water to the quinoxaline/Li triflate solution (Fig. S8b†) did not show a comparable effect on the quinoxaline voltammetry. Specifically, the magnitude of the reduction current and the shape of the voltammogram were not profoundly changed following water addition.
Our observation that trace water promotes the electrochemical properties of quinoxaline in some electrolyte but not others suggests that water itself does not directly electrochemically activate quinoxaline. Instead, the electrochemical behavior under study depends on a reaction between water and components of the LiBF4/PC electrolyte solution. Contamination by water is a well-known cause of degradation of LiBF4/PC electrolyte.21 Furthermore, the aging of as-prepared solutions of quinoxaline in LiBF4/PC, which contained 150 ppm of water, lead to increased reduction current as well, presumably due to the gradual degradation of the electrolyte following exposure to the water present in the quinoxaline stock (Fig. S9†). These results justify our application of computational studies to predict the interaction of quinoxaline with various LiBF4 degradation products, and the comparison of the calculated reduction potentials to the electrochemical data.
3.2 Computational analysis of quinoxaline–electrolyte interactions
Quantum chemical calculations are employed to better understand the quinoxaline–electrolyte interactions, particularly the role of BF3 in the observed electrochemical behavior. Being a Lewis acid, BF3 form adducts with basic quinoxalines which will, in turn, exhibit different electrochemical properties. A number of such scenarios are modeled for quinoxaline and 2,3,6-TMQ and the computed reduction potentials are compared to experimentally measured values. Broadly, the scenarios are: quinoxaline interacting with electrolyte, interacting with the salt and solvent (i.e., LiBF4/PC), and interacting with varying numbers of Lewis acid species (i.e., BF3, PF5). In Table 1, computed energetics (enthalpies and free energies) for the formation of likely adducts of quinoxaline and 2,3,6-TMQ and their reduction potentials are shown.| Entry | Complex | Lewis acid (LA) | Energetics (eV) | Reduction potential (V) | ||
|---|---|---|---|---|---|---|
| ΔH (soln) | ΔG (soln) | E (Red1) | E (Red2) | |||
| 1 | Q-LA | None | N/A | N/A | 1.55 | 1.01 |
| 2 | (PC)3–LiBF4 | 0.01 | 0.57 | 1.41 | 0.91 | |
| 3 | One BF3 | −0.81 | −0.25 | 2.40 | 1.80 | |
| 4 | Two BF3 | −1.35 | −0.24 | 3.20 | 2.77 | |
| 5 | Two BF3 (one side) | −0.85 | 0.11 | 2.18 | 2.18 | |
| 6 | Three BF3 | −1.38 | 0.11 | 3.20 | 3.01 | |
| 7 | Four BF3 | −1.41 | 0.47 | 3.20 | 3.07 | |
| 8 | One PF5 | −0.80 | −0.28 | 2.61 | 2.00 | |
| 9 | Two PF5 | −1.21 | −0.20 | 3.52 | 2.95 | |
| 10 | 2,3,6-TMQ-LA | None | N/A | N/A | 1.30 | 0.73 |
| 11 | One BF3 | −0.51 | 0.06 | 2.18 | 1.71 | |
| 12 | Two BF3 | −0.81 | 0.28 | 2.98 | 2.59 | |
| 13 | Three BF3 | −0.43 | 0.89 | 3.23 | 2.83 | |
| 14 | Four BF3 | 0.11 | 1.68 | 3.71 | 3.11 | |
| 15 | One PF5 | −0.37 | 0.17 | 2.40 | 1.90 | |
| 16 | Two PF5 | −0.43 | 0.38 | 2.92 | 2.92 | |
In Table 1, the entries 1 to 9 and 10 to 16 show different adducts of quinoxaline and 2,3,6-TMQ, respectively. The computed reduction potentials, corresponding to entries 1 to 7 of Table 1, are shown in Fig. 4 along with the schematic structures of these complexes. Entries denoted as exp a and exp b in Fig. 4 are from the experimental studies of Ames et al.9 and the present study. Ames et al. reported the quinoxaline reduction at −1.8 V vs. SCE (ca. 1.5 V vs. Li/Li+) in 0.1 M TEAP in dimethylformamide.9 This is qualitatively consistent with the experimental studies by Barqawi and Atfah who reported −1.62 V vs. SCE (ca. 1.7 V vs. Li/Li+) in 0.1 M TBAPF6 in acetonitrile (not shown).10 While neither of the previous authors reported a second electron transfer, cyclic voltammograms by Ames et al. show a lower potential second redox event which was not discussed.9
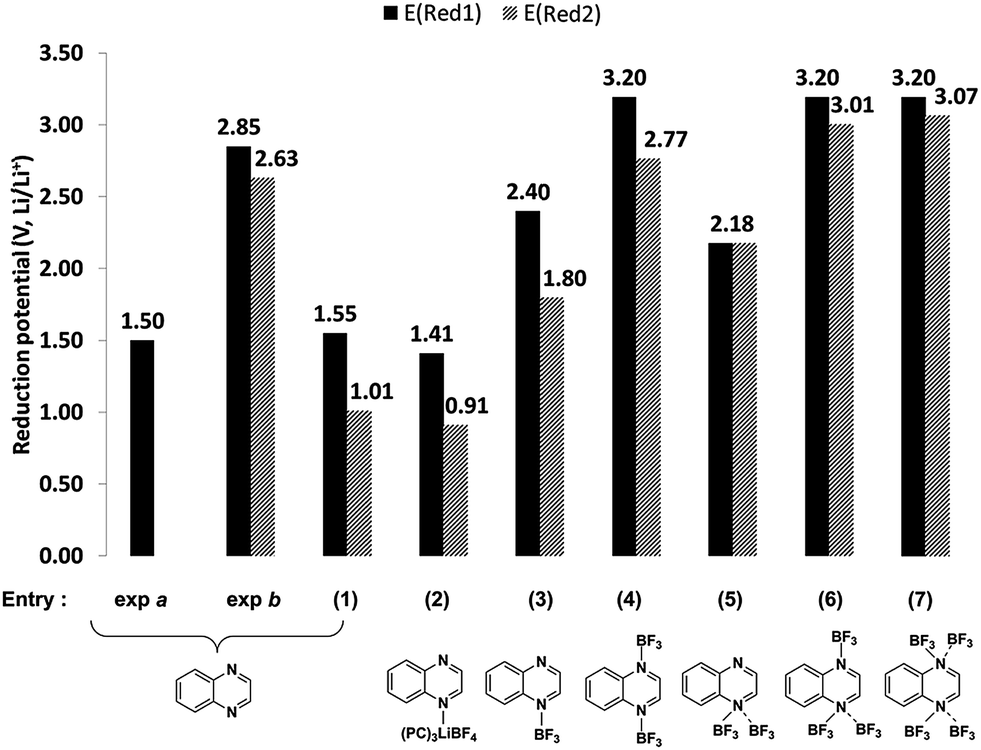 | ||
| Fig. 4 Comparison of computed first (E (Red1) and second reduction potentials (E (Red2) of quinoxaline (Q) with various adducts such as salt (LiBF4) and salt decomposition products (BF3). | ||
The computed reduction potentials of bare quinoxaline (entry 1 in Fig. 4) are 1.55 and 1.0 V respectively, consistent with previous experimental results.9,10 When explicit lithium salt and solvent (three PC molecules) were included (entry 2), computed reduction potentials are 1.41 and 0.91 V respectively, similar to the values for quinoxaline alone (entry 1). The computed reduction potentials of quinoxaline–BF3 adducts with one or more BF3 molecules are shown in entries 3–7. In general, the first reduction potentials are observed at higher potentials (by >1 V) when BF3 is present indicating a significant influence of BF3 towards the electrochemical behavior. The electron deficient Lewis acid, BF3, binds strongly with the nitrogen atoms of the quinoxalines and increases the electron affinity of the resulting Lewis acid-base adduct compared to the bare molecule. The computed electron affinities (EA) of quinoxaline and quinoxaline–2BF3 are 2.29 and 4.54 eV, respectively, indicating a significant increase in the electron affinity of the complex. In particular, the computed reduction potentials of quinoxaline–2BF3 (entry 4, Fig. 4), 3.20 and 2.77 V, are in reasonable agreement with the experimentally measured values (exp b, Fig. 4). Similarly, the 2,3,6-TMQ-2BF3 adduct (entry 12 in Table 1) is consistent with the experimentally observed reduction events of 2,3,6-TMQ in 0.2 M LiBF4/PC (Fig. 3a).
Ion-pairing effects on the electrochemical mechanism and reduction potentials of carbonyl-containing molecules have been examined in non-aqueous media by others.25 The formation of an ion-pair with the cation from the electrolyte stabilizes the electrochemically reduced anion, therefore shifting the electroreduction event towards positive potentials. We found that the calculated reduction potential of bare quinoxaline can be compared to previous results from Ames et al. in TBAPF6 in acetonitrile.9 In accordance with expectations from theory and the aforementioned previous studies, we observed that the electrochemical reduction potential of quinoxaline in LiBF4 and LiPF6 electrolyte was positively shifted compared to previous measurements. We found that quinoxaline did not appear electrochemically-active in TBABF4 or NaBF4 in propylene carbonate solution (ESI, Fig. S10†).
In terms of the BF3 binding with quinoxalines, from Table 1, the complexation enthalpies of one (entry 3), two (entry 4), three (entry 6), and four (entry 7) BF3 molecules with quinoxaline molecule is exothermic by 0.81, 1.35, 1.38, and 1.41 eV, respectively. Similarly, the complexation enthalpies of one (entry 11), two (entry 12), and three (entry 13) BF3 molecules with 2,3,6-TMQ are exothermic by 0.51, 0.81, and 0.43 eV, respectively. The binding of two molecules of BF3 (one molecule each with the nitrogen atom) appears optimal with the quinoxaline in solution (all optimized structures are shown in Fig. S11†). The binding of two BF3 to the same nitrogen atom (entry 5), a model which we note has unrealistic bonding, is energetically less favorable compared to two nitrogen atoms on opposite sides of the pyrazine heterocycle in the quinoxaline. Based on the calculations, Lewis acids such as BF3 bind more strongly with the quinoxaline than 2,3,6-TMQ due to the steric interaction from the methyl groups at positions 2 and 3 of the latter species. This is reflected in the complexation enthalpy of BF3 with quinoxalines and in the N–B bond length in the quinoxaline–BF3 complex. In their optimized geometries, shown in Fig. 5, the N–B bond lengths are 1.72 Å and 1.75 Å for quinoxaline and 2,3,6-TMQ based complexes, respectively. This is also consistent with a relatively stronger coordination of quinoxaline with the BF3 than the TMQ.
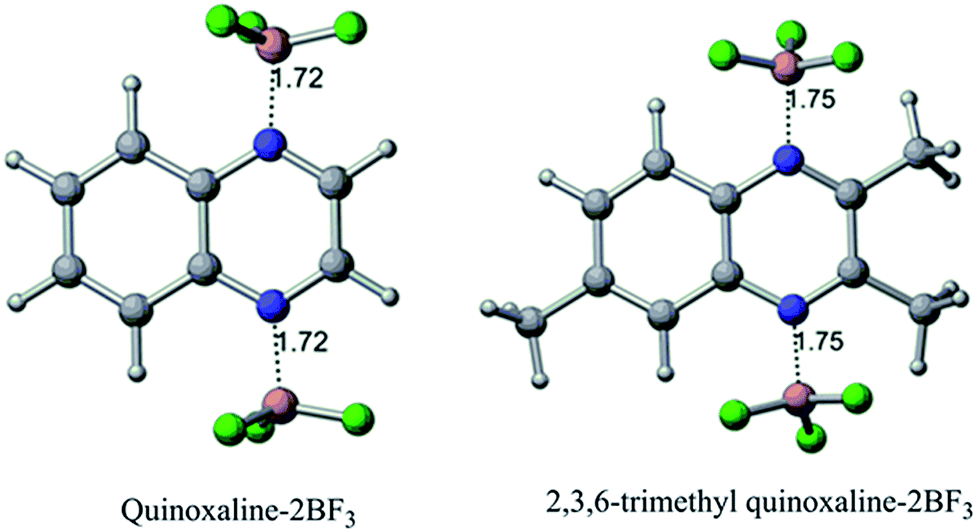 | ||
| Fig. 5 Optimized structures of quinoxaline and 2,3,6-TMQ complexed with two BF3 molecules computed at the B3LYP/6-31+G(d) level of theory. Selected bond lengths, in Å, between nitrogen and boron at the neutral state are also shown. | ||
Calculations presented in Table 1 suggest that the binding of BF3 with quinoxaline and TMQ is different and the extent of binding may affect the redox properties of the molecule. To understand the ability of other quinoxaline derivatives to become active in the presence of BF3, we have computed enthalpies and free energies of complexation of BF3 to seven selected quinoxaline derivatives, which are shown in Table 2 (entries 1 to 11). The binding of BF3 molecules with quinoxaline containing electron withdrawing substituents such as chloro (entries 1 & 2) or trifluoromethyl (entry 3) are not energetically favorable, suggesting that these molecules are less likely to exhibit electrochemical behavior similar to quinoxaline or 2,3,6-TMQ, which is consistent with the results reported here and in earlier work.8 Quinoxaline with acetyl (entries 4,5), phenyl (entries 6,7), or etheric groups (entries 8 to 11) are likely to show augmented electrochemical properties in the presence of LiBF4 due to the binding of BF3 molecules. The enthalpy of complexation of these molecules (entries 4 to 11) with BF3 is not as strong as either quinoxaline or 2,3,6-TMQ indicating that the effect of salt or salt decomposition products in enhancing the electrochemical properties are minimal for these molecules.
| Entry | Quinoxaline derivative | Lewis acid (LA) | Energetics (eV) | |
|---|---|---|---|---|
| ΔH (soln) | ΔG (soln) | |||
| 1 | 2,3-Dichloroquinoxaline | One BF3 | −0.06 | 0.46 |
| 2 | Two BF3 | 0.13 | 0.9 | |
| 3 | 2,3-Di-trifluromethylquinoxaline | Two BF3 | 0.1 | 0.87 |
| 4 | 2-Acetylquinoxaline | One BF3 | −0.36 | 0.2 |
| 5 | 2,3-Diacetylquinoxaline | Two BF3 | −0.43 | 0.63 |
| 6 | 2,3-Diphenylquinoxaline | One BF3 | −0.42 | 0.14 |
| 7 | Two BF3 | −0.59 | 0.51 | |
| 8 | 2,3-Dimethoxyquinoxaline | One BF3 | −0.44 | 0.12 |
| 9 | Two BF3 | −0.38 | 0.5 | |
| 10 | 2-Ethoxymethoxyquinoxaline | One BF3 | −0.52 | 0.07 |
| 11 | Two BF3 | −0.48 | 0.64 | |
3.3 Validation of quinoxaline–2BF3 complex activity
Having predicted the formation of an electro-active quinoxaline–BF3 adduct through electrochemical experiments and quantum calculations, we sought to further verify the presence of this species by directly synthesizing and testing a quinoxaline (bis)trifluoroborane ((2BF3)Q) complex. The complex was synthesized as described above, and evaluated in Li triflate/PC electrolyte, in which quinoxalines were previously shown to be inactive (see Fig. 1). Fig. 6 shows the cyclic voltammograms for 5 mM (2BF3)Q in 0.2 M Li triflate/PC as compared to 5 mM quinoxaline (denoted Q in the figure) in 0.2 M LiBF4/PC. The (2BF3)Q demonstrates an order of magnitude increase in current as compared to quinoxaline at the same concentration. The reduction waves around 3.2 V and 2.7 V are in good agreement with the range of computed reduction potentials for various (2BF3)n-adducted quinoxaline structures (Table 1 & Fig. 4). The presence of more than 2 reduction waves may indicate the presence of multiple electro-active species.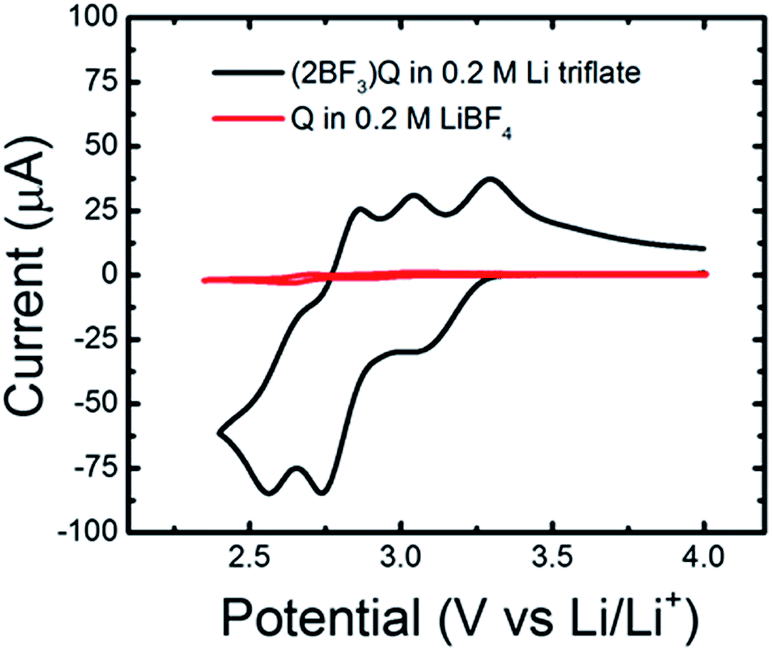 | ||
| Fig. 6 Cyclic voltammetry of 5 mM quinoxaline (bis)trifluoroborane ((2BF3)Q) and 5 mM quinoxaline (Q) in 0.2 M Li triflate/PC and 0.2 M LiBF4/PC respectively. Experiments were performed at a 20 mV s−1 scan rate. | ||
In addition to CV analysis, we performed bulk electrolysis experiments on the two electrolyte solutions, 5 mM (2BF3)Q in 0.2 M Li triflate/PC and 5 mM quinoxaline in 0.2 M LiBF4/PC. The solutions were cycled at 5 mA between 2.4 V and 4.0 V using constant-current chronopotentiometry. Note that these are the same potential limits used for comparison of CV data in Fig. 6. Identical bulk electrolysis conditions were used as well (solution volume, stir rate). Fig. 7 presents capacity curves constructed from the cycling data for eight charge–discharge cycles, with the last four cycles indicated by dotted lines. Considering the possibility that 2 electrons may transfer to each molecule (n = 2e−) in accordance with the DFT studies in the previous sections, the maximum theoretical capacity at 100% state-of-charge (SOC) is 482 mA h. During the first charging cycle, the quinoxaline-containing solution achieved a capacity of only 10 mA h, whereas the (2BF3)Q-containing solution reached a capacity of 260 mA h. These values correspond respectively to 2% and 54% of the maximum theoretical state-of-charge (SOC), and also indicate a 26-fold increase the amount of electrochemically-active charge-storage material for the (2BF3)Q species. These results are in agreement with results from cyclic voltammetry presented in Fig. 6 which also showed significant enhancement in the concentration of electrochemically-active species. The charging and discharging capacities increased by about 10% of the maximal theoretical SOC over the course of cycling for both the quinoxaline and (2BF3)Q species. Interestingly, while the SOC for the quinoxaline-containing solution never exceeded 10% of the theoretical maximum SOC for an n = 2e− reduction, the SOC obtained for the (2BF3)Q-containing solution was approximately 50% of the theoretical maximum SOC for an n = 2e−, or alternatively 100% SOC for an n = 1e− reduction. Unexpectedly, coulombic efficiencies in excess of 100% were observed for both molecules and the discharge capacity also increased with time. Preliminary NMR analysis of the cycled quinoxaline solutions was inconclusive due to the presence of a large number of new peaks. This indicates that bulk electrolysis cycling generates electrochemically active side-products. In addition, we observed a gradual and irreversible change in solution color, from clear to dark blue for quinoxaline and clear to dark red for (2BF3)Q over the course of cycling, also pointing to structural evolution of quinoxaline. Therefore, further exploration of quinoxaline stability and the charge mechanisms are imperative to determining the fitness of quinoxalines as charge-storage materials in non-aqueous organic RFBs but are beyond the scope of the current paper. We aim to address these topics more specifically in subsequent studies.
 | ||
| Fig. 7 Capacity curves from the constant-current chronopotentiometry bulk electrolysis cycling of 5 mM quinoxaline in 0.2 M LiBF4/PC (top) and 5 mM 2(BF3)Q in 0.2 M Li triflate/PC (bottom). The charging and discharging current was 5 mA. 8 charge–discharge cycles are shown, with dotted lines representing the last 4 cycles. | ||
4. Conclusions
We have investigated the impact of electrolyte composition on quinoxalines in PC solvent using electrochemical methods and quantum chemical simulations. Lewis acid salt decomposition products (i.e., BF3 from LiBF4) are found to bind strongly with the nitrogen atoms of quinoxaline and increases electron affinity and thus raises the reduction potential. The observed activity in LiBF4 (or LiPF6)-based electrolytes, between 2.4–3.2 V vs. Li/Li+, is not due to quinoxaline alone but rather an electro-active quinoxaline–BF3 complex. This is approximately 1–1.5 V higher than previous observations of quinoxaline electrochemical behavior in non-aqueous media. The salt effect is further validated by synthesizing and testing a quinoxaline–2BF3 complex, an optimal configuration according to quantum chemical calculations. As compared to quinoxaline in LiBF4-based electrolyte, synthesized (2BF3)Q demonstrates up to a 26-fold increase in charging capacity, using an electrolyte in which bare quinoxaline is inactive.These results advance our understanding of the impact of electrolyte decomposition products on the electrochemical behavior of quinoxaline in propylene carbonate. The insight regarding electrochemical activation via BF3 adduct-forming may lead to new classes of redox-active materials for non-aqueous flow battery applications. In continuing studies, we will employ more advanced electrochemical methods, including in situ spectroscopy, to focus on the structural evolution of quinoxaline during and after electrochemical reduction, as well as the precise role of the solvent in directing electrochemical properties.
Acknowledgements
This work was supported as part of the Joint Center for Energy Storage Research, an Energy Innovation Hub funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences. We gratefully acknowledge the computing resources provided on “Blues,” a 320-node computing cluster operated by the Laboratory Computing Resource Center at Argonne National Laboratory. Use of the Center for Nanoscale Materials was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract no. DE-AC02-06CH11357.References
- J. Eyer and G. Gorey, Energy Storage for the Electricity Grid: Benefits and Market Potential Assessment Guide, Sandia Report SAND2010-0815, 2010 Search PubMed.
- (a) D. Rastler, Electricity Energy Storage Options: A White Paper Primer on Applications, Costs and Benefits, EPRI Technical Update 1020676, 2010 Search PubMed; (b) Z. G. Yang, J. L. Zhang, M. C. W. Kintner-Meyer, X. C. Lu, D. W. Choi, J. P. Lemmon and J. Liu, Electrochemical Energy Storage for Green Grid, Chem. Rev., 2011, 111(5), 3577–3613 CrossRef CAS PubMed; (c) W. Wang, Q. T. Luo, B. Li, X. L. Wei, L. Y. Li and Z. G. Yang, Recent Progress in Redox Flow Battery Research and Development, Adv. Funct. Mater., 2013, 23(8), 970–986 CrossRef CAS; (d) M. Skyllas-Kazacos, M. H. Chakrabarti, S. A. Hajimolana, F. S. Mjalli and M. Saleem, Progress in Flow Battery Research and Development, J. Electrochem. Soc., 2011, 158(8), R55–R79 CrossRef CAS PubMed; (e) B. Dunn, H. Kamath and J. M. Tarascon, Electrical Energy Storage for the Grid: A Battery of Choices, Science, 2011, 334(6058), 928–935 CrossRef CAS PubMed.
- A. Z. Weber, M. M. Mench, J. P. Meyers, P. N. Ross, J. T. Gostick and Q. H. Liu, Redox flow batteries: a review, J. Appl. Electrochem., 2011, 41(10), 1137–1164 CrossRef CAS.
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, A metal-free organic-inorganic aqueous flow battery, Nature, 2014, 505(7482), 195–198 CrossRef CAS PubMed.
- (a) Y. Matsuda, K. Tanaka, M. Okada, Y. Takasu, M. Morita and T. Matsumurainoue, A Rechargeable Redox Battery Utilizing Ruthenium Complexes with Non-Aqueous Organic Electrolyte, J. Appl. Electrochem., 1988, 18(6), 909–914 CrossRef CAS; (b) Q. H. Liu, A. E. S. Sleightholme, A. A. Shinkle, Y. D. Li and L. T. Thompson, Non-aqueous vanadium acetylacetonate electrolyte for redox flow batteries, Electrochem. Commun., 2009, 11(12), 2312–2315 CrossRef CAS PubMed; (c) J. Mun, M. J. Lee, J. W. Park, D. J. Oh, D. Y. Lee and S. G. Doo, Non-Aqueous Redox Flow Batteries with Nickel and Iron Tris(2,2 '-bipyridine) Complex Electrolyte, Electrochem. Solid-State Lett., 2012, 15(6), A80–A82 CrossRef CAS PubMed.
- T. M. Anderson, D. Ingersoll, A. J. Rose, C. L. Staiger and J. C. Leonard, Synthesis of an ionic liquid with an iron coordination cation, Dalton Trans., 2010, 39(37), 8609–8612 RSC.
- (a) Z. Li, S. Li, S. Q. Liu, K. L. Huang, D. Fang, F. C. Wang and S. Peng, Electrochemical Properties of an All-Organic Redox Flow Battery Using 2,2,6,6-Tetramethyl-1-Piperidinyloxy and N-Methylphthalimide, Electrochem. Solid-State Lett., 2011, 14(12), A171–A173 CrossRef CAS PubMed; (b) W. Wang, W. Xu, L. Cosimbescu, D. W. Choi, L. Y. Li and Z. G. Yang, Anthraquinone with tailored structure for a nonaqueous metal-organic redox flow battery, Chem. Commun., 2012, 48(53), 6669–6671 RSC; (c) W. D. Zhou, K. Hernandez-Burgos, S. E. Burkhardt, H. L. Qian and H. D. Abruna, Synthesis and Electrochemical and Computational Analysis of Two New Families of Thiophene-Carbonyl Molecules, J. Phys. Chem. C, 2013, 117(12), 6022–6032 CrossRef CAS.
- F. R. Brushett, J. T. Vaughey and A. N. Jansen, An All-Organic Non-aqueous Lithium-Ion Redox Flow Battery, Adv. Energy Mater., 2013, 2(11), 1390–1396 CrossRef.
- J. R. Ames, M. A. Houghtaling and D. L. Terrian, Cyclic voltammetry of some quinoxaline di-N-oxides and quinoxalines in dimethylformamide, Electrochim. Acta, 1992, 37(8), 1433–1436 CrossRef CAS.
- K. R. Barqawi and M. A. Atfah, A cyclic voltammetric study of some quinoxaline di-n-oxides and quinoxalines in acetonitrile: substituent effect on the cathodic reduction, Electrochim. Acta, 1987, 32(4), 597–599 CrossRef CAS.
- G. Angulo, J. Dobkowski, A. Kapturkiewicz and K. Maciolek, Photophysics and electrochemistry of quinoxaline chromophores decorated with thiophene or furane subunits, J. Photochem. Photobiol., A, 2010, 213(23), 101–106 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396 CrossRef CAS PubMed.
- A. A. Isse and A. Gennaro, Absolute Potential of the Standard Hydrogen Electrode and the Problem of Interconversion of Potentials in Different Solvents, J. Phys. Chem. B, 2010, 114(23), 7894–7899 CrossRef CAS PubMed.
- (a) C. P. Kelly, C. J. Cramer and D. G. Truhlar, Aqueous Solvation Free Energies of Ions and Water Clusters Based on an Accurate Value for the Absolute Aqueous Solvation Free Energy of the Proton, J. Phys. Chem. B, 2006, 110(32), 16066–16081 CrossRef CAS PubMed; (b) S. Bhattacharyya, M. T. Stankovich, D. G. Truhlar and J. Gao, Combined Quantum Mechanical and Molecular Mechanical Simulations of One- and Two-Electron Reduction Potentials of Flavin Cofactor in Water, Medium-Chain Acyl-CoA Dehydrogenase, and Cholesterol Oxidase, J. Phys. Chem. A, 2007, 111(26), 5729–5742 CrossRef CAS PubMed; (c) R. S. Assary, L. A. Curtiss and J. S. Moore, Toward a Molecular Understanding of Energetics in Li–S Batteries Using Nonaqueous Electrolytes: A High-Level Quantum Chemical Study, J. Phys. Chem. C, 2014 Search PubMed.
- J. J. Guerard and J. S. Arey, Critical Evaluation of Implicit Solvent Models for Predicting Aqueous Oxidation Potentials of Neutral Organic Compounds, J. Chem. Theory Comput., 2013, 9(11), 5046–5058 CrossRef CAS.
- (a) J. Moens, P. Geerlings and G. Roos, A Conceptual DFT Approach for the Evaluation and Interpretation of Redox Potentials, Chem.–Eur. J., 2007, 13(29), 8174–8184 CrossRef CAS PubMed; (b) O. Borodin, W. Behl and T. R. Jow, Oxidative Stability and Initial Decomposition Reactions of Carbonate, Sulfone, and Alkyl Phosphate-Based Electrolytes, J. Phys. Chem. C, 2013, 117(17), 8661–8682 CrossRef CAS; (c) J. M. Vollmer, A. Kandalam, P. Zapol, L. A. Curtiss, C. H. Chen, D. R. Vissers and K. Amine, Prediction of reduction potentials with quantum chemical methods, Batteries and Supercapacitors 2003, pp. 389–394 Search PubMed; (d) J. M. Vollmer, L. A. Curtiss, D. R. Vissers and K. Amine, Reduction Mechanisms of Ethylene, Propylene, and Vinylethylene Carbonates: A Quantum Chemical Study, J. Electrochem. Soc., 2004, 151(1), A178–A183 CrossRef CAS PubMed; (e) C. P. Kelly, C. J. Cramer and D. G. Truhlar, Single-Ion Solvation Free Energies and the Normal Hydrogen Electrode Potential in Methanol, Acetonitrile, and Dimethyl Sulfoxide, J. Phys. Chem. B, 2006, 111(2), 408–422 CrossRef PubMed; (f) L. Xing, O. Borodin, G. D. Smith and W. Li, Density Functional Theory Study of the Role of Anions on the Oxidative Decomposition Reaction of Propylene Carbonate, J. Phys. Chem. A, 2011, 115(47), 13896–13905 CrossRef CAS PubMed; (g) R. L. Wang, C. Buhrmester and J. R. Dahn, Calculations of Oxidation Potentials of Redox Shuttle Additives for Li-Ion Cells, J. Electrochem. Soc., 2006, 153(2), A445–A449 CrossRef CAS PubMed; (h) R. S. Assary, L. A. Curtiss, P. C. Redfern, Z. Zhang and K. Amine, Computational Studies of Polysiloxanes: Oxidation Potentials and Decomposition Reactions, J. Phys. Chem. C, 2011, 115(24), 12216–12223 CrossRef CAS.
- A. Z. Szarka, L. A. Curtiss and J. R. Miller, Calculation of temporary anion states using density functional theory, Chem. Phys., 1999, 246(1–3), 147–155 CrossRef CAS.
- (a) S. E. Burkhardt, M. A. Lowe, S. Conte, W. D. Zhou, H. L. Qian, G. G. Rodriguez-Calero, J. Gao, R. G. Hennig and H. D. Abruna, Tailored redox functionality of small organics for pseudocapacitive electrodes, Energy Environ. Sci., 2012, 5(5), 7176–7187 RSC; (b) L. Xing and O. Borodin, Oxidation induced decomposition of ethylene carbonate from DFT calculations - importance of explicitly treating surrounding solvent, Phys. Chem. Chem. Phys., 2012, 14(37), 12838–12843 RSC.
- D. R. Martin, C. M. Merkel, C. B. Drake, J. U. Mondal and J. B. Iwamoto, Tribromo- and trifluoroborane adducts of some pyrazines, Inorg. Chim. Acta, 1985, 97(2), 189–193 CrossRef CAS.
- K. Xu, Nonaqueous Liquid Electrolytes for Lithium-Based Rechargeable Batteries, Chem. Rev., 2004, 104(10), 4303–4418 CrossRef CAS.
- T. Pilz, T. Schleid and M. Jansen, The Lithium Fluoride Tetrafluoroborate Li2F(BF4), Z. Anorg. Allg. Chem., 2013, 639(14), 2555–2557 CrossRef CAS.
- K. Tasaki, K. Kanda, S. Nakamura and M. Ue, Decomposition of LiPF6 and Stability of PF5 in Li-Ion Battery Electrolytes: Density Functional Theory and Molecular Dynamics Studies, J. Electrochem. Soc., 2003, 150(12), A1628–A1636 CrossRef CAS PubMed.
- (a) W. T. Li, C. Campion, B. L. Lucht, B. Ravdel, J. DiCarlo and K. M. Abraham, Additives for stabilizing LiPF6-based electrolytes against thermal decomposition, J. Electrochem. Soc., 2005, 152(7), A1361–A1365 CrossRef CAS PubMed; (b) S. S. Zhang, K. Xu and T. R. Jow, A thermal stabilizer for LiPF6-based electrolytes of Li-ion cells, Electrochem. Solid-State Lett., 2002, 5(9), A206–A208 CrossRef CAS PubMed.
- M. Takahashi and M. Ito, Surface-enhanced Raman spectra of quinoxaline adsorbed at a silver electrode, Chem. Phys. Lett., 1984, 103(6), 512–516 CrossRef CAS.
- K. Hernandez-Burgos, G. G. Rodríguez-Calero, W. Zhou, S. E. Burkhardt and H. D. Abruña, Increasing the Gravimetric Energy Density of Organic Based Secondary Battery Cathodes Using Small Radius Cations (Li+, and Mg2+), J. Am. Chem. Soc., 2013, 135(39), 14532 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Computed reduction potentials quinoxaline in various dielectric mediums, electrochemical data for quinoxaline in the presence of TBABF4 salt, electrochemical data for quinoxaline–BF3·OEt2 in PC without supporting salt, optimized structure of quinoxaline–BF3 complexes. See DOI: 10.1039/c5ra00137d |
| ‡ Current address: Schulich Faculty of Chemistry, Technion – Israel Institute of Technology, Technion city, Haifa, 320008, Israel. |
| This journal is © The Royal Society of Chemistry 2015 |