
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluation of undoped and M-doped TiO2, where M = Sn, Fe, Ni/Nb, Zr, V, and Mn, for lithium-ion battery applications prepared by the molten-salt method
M. V.
Reddy
*ab,
Neeraj
Sharma
*c,
Stefan
Adams
*a,
R. Prasada
Rao
a,
Vanessa K.
Peterson
d and
B. V. R.
Chowdari
b
aDepartment of Materials Science & Engineering, National University of Singapore, Singapore 117576. E-mail: msemvvr@nus.edu.sg; phymvvr@nus.edu.sg; mseasn@nus.edu.sg
bDepartment of Physics, National University of Singapore, Singapore 117542
cSchool of Chemistry, The University of New South Wales, Sydney 2052, NSW, Australia. E-mail: neeraj.sharma@unsw.edu.au
dAustralian Nuclear Science and Technology Organisation, Kirrawee DC 2232, NSW, Australia
First published on 16th March 2015
Abstract
The molten-salt method was used to synthesize a series of transition-metal containing titanium dioxides. Some of the transition metals were found to substitute into the TiO2 lattice, such as (Ti0.9Fe0.1)O2, (Ti0.9Zr0.1)O2, (Ti0.9V0.1)O2, and (Ti0.9Mn0.1)O2, while others were formed as composite electrodes (in addition to relatively minor substitutions), namely 0.1SnO2–0.9TiO2 and 0.05NiO–0.1Nb2O5–0.9TiO2. Although identical synthesis-conditions were used the different transition metals yielded different phases. A comparative study of the electrodes relating surface area and composition (via X-ray photoelectron spectroscopy, XPS), and electrochemical behaviour is presented in this work. Among the substituted single phase electrodes, (Ti0.9Zr0.1)O2 exhibited the best reversible capacity of ∼160 mA h g−1, at the end of the 60th cycle in the voltage range 1.0–2.6 V, with a capacity fade of 24% from the 2nd to the 60th cycle. Among the composite electrodes, 0.05NiO–0.1Nb2O5–0.9TiO2 shows the best performance which is comparable to pure TiO2 but with a slower capacity-fade on extended cycling. The worst performing electrode is (Ti0.9V0.1)O2 with a reversible capacity of only ∼70 mA h g−1 at the end of 70 cycles with a current density of 130 mA g−1 in the voltage range 1.0–2.6 V and a capacity drop of 52% from the 2nd to the 70th cycle. The composite 0.1SnO2–0.9TiO2 features the highest irreversible capacity-loss. Zr-substitution into TiO2 gives the best electrochemical performance.
1. Introduction
Studies on anode materials for lithium-ion batteries have recently shifted towards transition-metal oxides. Transition-metal oxides such as TiO2 are considered as potential candidates for high-power anodes in lithium-ion batteries because of their low cost, availability in significant quantities, and eco-friendliness.1,2 TiO2 meets these favourable properties but exhibits higher lithium-insertion potentials relative to the commonly-used graphitic anodes.3–5 Although, high potentials for lithium-insertion reduce battery voltage, they alleviate the hazard of lithium plating on the anode, a common safety problem limiting high rate charging of graphitic anodes.6,7 Similar to the attractive Li4Ti5O128–12 anode which exhibits a relatively minor volume expansion (<1%) with lithium insertion/extraction, various polymorphs of TiO2 have been investigated and shown to be attractive lithium-hosts.13 Of these, anatase TiO2 is the most electroactive insertion-host for lithium,13–15 however, significant capacity fade is found in anatase TiO2-containing batteries.The capacity fade can be suppressed by using aliovalent and isovalent transition-metal doping at the titanium site or nitrogen doping at the oxygen site. This leads to formulations of Ti1−xMxO2, where M is a transition-metal cation. In the case of isovalent M charge balance is not affected, while for aliovalent doping, either oxygen vacancies (in the case of M3+ dopants) or a reduction in the titanium oxidation-state from 4+ to 3+ (in the case of supervalent doping with M5+) are expected to maintain charge balance. The advantage of supervalent doping compared to isovalent doping is that the conventional titanium oxidation state of 4+ found in TiO2 is replaced with a combination of Ti3+ and Ti4+ which will enhance electronic conductivity of the anode, in addition to the direct effect of the supervalent cation. Using a lower oxidation-state cation can produce a similar enhancement of electronic conductivity by virtue of different oxidation states in one material. The cost of the electronic-conductivity enhancement by doping is the loss of potential Ti4+ that can be reduced to Ti3+ during the insertion of lithium, i.e. a slight reduction in capacity. This compromise in battery performance between electrical conductivity and the quantity of lithium inserted/extracted requires input from the battery manufacturer or researcher.
Not all transition metals form doped TiO2 when synthesized under the same conditions. In these cases, multiphase composite-electrodes are formed where the major component is TiO2 (or a low-doped equivalent) and a minor component which is predominantly composed of the dopant oxide. Although some mixing can occur, e.g. some transition-metal substituted into TiO2 or Ti substituted into the transition-metal oxide, determining the extent of such doping is difficult due to the small quantities of transition-metals and therefore the exceptionally-small amount of mixing that occurs. The composite can be identified by the presence of both TiO2 and the transition-metal oxide phases in diffraction patterns. They can be written as xMOy·1−xTiO2, where M is the transition metal, y is related to the oxidation state of the transition metal M, and x is the ratio between the components of the composite. The performance and properties of batteries made of such composite electrodes can be related to the major and minor components of the composite. It has been shown for an idealised Li4Ti5O12/TiO2 composite, that both components are electrochemically active at specific potentials.11
The molten-salt method is one of the most versatile and simple methods to prepare various metal oxides16–20 with controlled morphology and tailored physical and electrochemical properties.21,22 In this work, the molten-salt method under specific conditions is used to prepare transition-metal doped titanium dioxides, Ti1−xMxO2, and composite compounds, xMOy·1−xTiO2, to compare the electrochemical properties with those of anatase TiO2 to determine whether doping can improve electrochemical performance. The choice of transition metal determines whether substituted or composite electrodes are formed and for each series, the best and worst performing electrodes are compared.
2. Experimental
Titanium dioxides of the following Ti1−x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mx composition, Ti0.9Sn0.1, Ti0.9Fe0.1, Ti0.9Zr0.1, Ti0.9V0.1, Ti0.9Mn0.1, and Ti0.85Ni0.05Nb0.1, were prepared using the molten-salt method. Ti0.9Sn0.1 was synthesized using TiOSO4 x H2SO4 x H2O (Sigma-Aldrich, 99%) and SnCl4·4H2O (Riedel-de Haën, 98%) in a mole ratio of 0.9:0.1 using 0.5 M LiNO3 (Alfa Aesar, 68.95%) and 0.5 M KNO3 (Merck, 99.0%) to facilitate the method. These compounds were thoroughly mixed and placed in an alumina crucible and heated to 410 °C for 1 h. To prepare Ti0.9Fe0.1, Ti0.9Zr0.1, Ti0.9V0.1, and Ti0.9Mn0.1, SnCl4·4H2O was replaced by FeSO4·7H2O (Aldrich, 99%), Zr(SO4)2 (Merck, purity 99.5%), VOSO4 (Merck, purity 99%), and MnSO4·4H2O (Alfa Aesar, 99%), respectively. For Ti0.85Ni0.05Nb0.1, SnCl4·4H2O was replaced by Ni(NO3)2·6H2O and Nb2O5 in a ratio of TiOSO4:Ni(NO3)2·6H2O:Nb2O5 = 0.85:0.05:0.1. Analogous mixing and heating protocols to that used in the preparation of Ti0.9Sn0.1O2 were used to synthesize the other compounds. After furnace heating, the obtained materials were dissolved in de-ionized water to remove the Li and K salts, filtered, and finally dried overnight in a vacuum oven at 80 °C. The compounds were stored in a desiccator.
:Mx composition, Ti0.9Sn0.1, Ti0.9Fe0.1, Ti0.9Zr0.1, Ti0.9V0.1, Ti0.9Mn0.1, and Ti0.85Ni0.05Nb0.1, were prepared using the molten-salt method. Ti0.9Sn0.1 was synthesized using TiOSO4 x H2SO4 x H2O (Sigma-Aldrich, 99%) and SnCl4·4H2O (Riedel-de Haën, 98%) in a mole ratio of 0.9:0.1 using 0.5 M LiNO3 (Alfa Aesar, 68.95%) and 0.5 M KNO3 (Merck, 99.0%) to facilitate the method. These compounds were thoroughly mixed and placed in an alumina crucible and heated to 410 °C for 1 h. To prepare Ti0.9Fe0.1, Ti0.9Zr0.1, Ti0.9V0.1, and Ti0.9Mn0.1, SnCl4·4H2O was replaced by FeSO4·7H2O (Aldrich, 99%), Zr(SO4)2 (Merck, purity 99.5%), VOSO4 (Merck, purity 99%), and MnSO4·4H2O (Alfa Aesar, 99%), respectively. For Ti0.85Ni0.05Nb0.1, SnCl4·4H2O was replaced by Ni(NO3)2·6H2O and Nb2O5 in a ratio of TiOSO4:Ni(NO3)2·6H2O:Nb2O5 = 0.85:0.05:0.1. Analogous mixing and heating protocols to that used in the preparation of Ti0.9Sn0.1O2 were used to synthesize the other compounds. After furnace heating, the obtained materials were dissolved in de-ionized water to remove the Li and K salts, filtered, and finally dried overnight in a vacuum oven at 80 °C. The compounds were stored in a desiccator.
Structural characterization of the compounds was undertaken by X-ray powder diffraction (XRD) using a Siemens D5005 diffractometer equipped with CuKα radiation. Rietveld analysis was undertaken using the TOPAS software suite.23 Neutron powder-diffraction (NPD) data were collected for the Ti0.9Mn0.1O2 sample using the high-intensity powder diffractometer, WOMBAT,24 at the Open Pool Australian Light-water (OPAL) reactor facility at the Australian Nuclear Science and Technology Organisation (ANSTO).25 The sample was placed in a 6 mm vanadium can and data collected for 5 min between 30 ≤ 2θ ≤ 130° using λ = 1.5407(1) Å, with the wavelength determined using the NIST 676 Al2O3 standard reference material. WOMBAT is equipped with a two-dimensional detector allowing data to be collected over the entire 2θ-range in one acquisition. Rietveld refinements were carried out using GSAS25 with the EXPGUI26 interface. Absorption was accounted for in the Rietveld model.27,28 The morphology of the powders was examined using transmission electron-microscopy (TEM). Local information concerning the bonding environment of pure and transition-metal containing TiO2 was determined using Raman spectroscopy. The surface area of the as-synthesized samples was measured using the Brunauer–Emmett–Teller (BET) method. X-ray photoelectron spectroscopy (XPS) of the bare and doped compounds was undertaken using an AXIS ultra DLD spectrometer (Kratos Analytica) with monochromatic AlKα radiation. The survey spectra were obtained in the range 0–1200 eV. Charge referencing was carried out against adventitious carbon C (C1s binding energy = 284.6 eV). Casa XPS software was used to analyse XPS spectral data.
Electrodes for electrochemical studies were made using a mixture of the active, as-synthesized materials, Super P carbon, and polyvinylidene fluoride binder (PVDF, Kynar 2801) in the weight ratio of 70:15:15. A thick slurry of this mixture was made using N-methyl pyrrolidone (NMP, Alfa Aesar) as the solvent and coated onto a copper foil by the doctor-blade technique.29 The geometrical area of the electrode was 2.0 cm2 and weight of the active material was 4–5 mg. Coin-cell fabrication was undertaken in a glovebox using lithium metal as the counter electrode, LiPF6 in dimethyl carbonate and ethylene carbonate (1:1 mol%) as the electrolyte, and the electrode. These were then galvanostatically cycled between 1.0 and 2.6 V at a current rate of 130 mA g−1, using a potentiostat/galvanostat (Bitrode battery tester) at room temperature. A Macpile system was used for cyclic voltammetry studies. Li metal was used as the counter and reference electrode in the range 1.0–2.6 V at ambient temperature, and the scan rate was 0.058 mV s−1 for up to 6 cycles. More details on fabrication and instrumentation are reported in a previous study.30,31
3. Results and discussion
The Rietveld refinement profiles using the XRD data of the synthesized samples are shown in Fig. 1 and NPD data of a selected compound (Ti0.9Mn0.1)O2 are shown in Fig. 2. Data for samples with the transition-metal dopants Fe, Zr, Mn, and V indicate successful doping into the single-phase compound Ti1−xMxO2−y adopting the tetragonal anatase structure (space group I41/amd). Note, in the above formula y is a possible change in the oxygen content required for charge balance on aliovalent doping. The remaining transition-metal samples form composite (multiphase) materials with xM′Oy·1−xTiO2, where M′ represents the transition metals Sn and Nb/Ni and y depends on the oxidation state of the transition metal. In these cases the second phase is cassiterite SnO2 or Nb2O5. In the case of the co-doped Nb/Ni compound, the possibility exists of relatively minor Ni incorporation into the anatase structure while Nb forms a secondary phase, however, the details are below the limit of detection of the techniques used. The lattice parameters of the major anatase-type TiO2 phase in each case are shown in Table 1.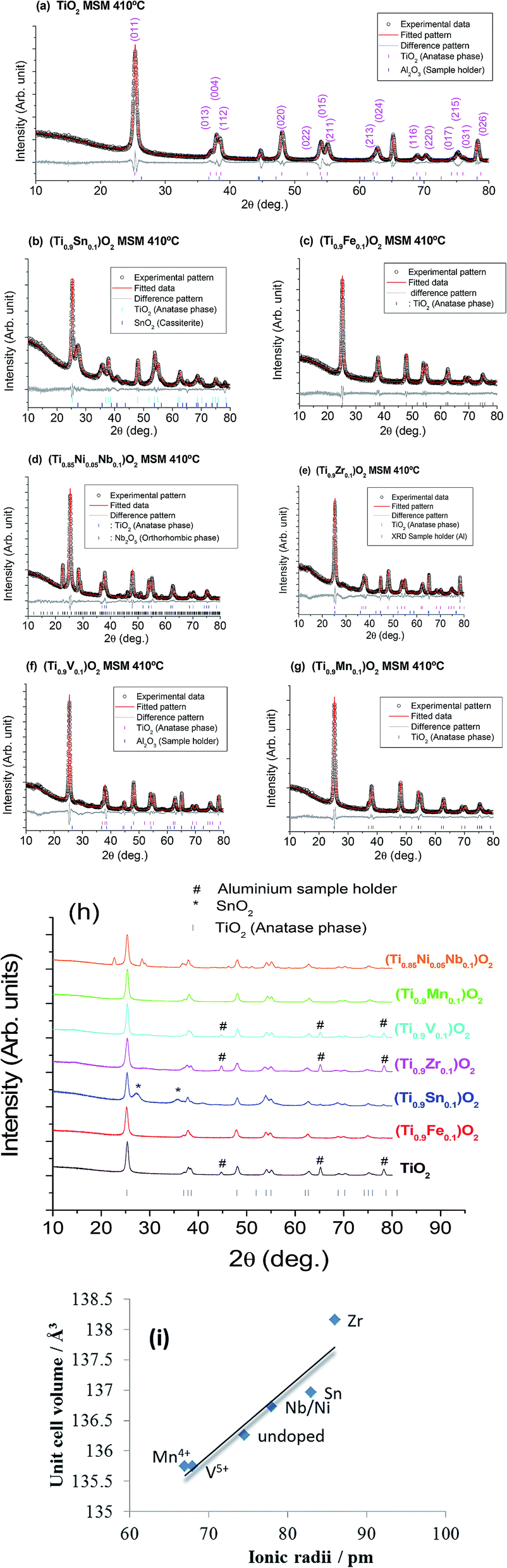 | ||
| Fig. 1 Observed (black crosses) X-ray powder diffraction data and Rietveld refined fits (red) of (a) TiO2 with reflections labelled, (b) 0.1SnO2·0.9TiO2, (c) (Ti0.9Fe0.1)O2, (d) 0.1NiO·0.05Nb2O5·0.85TiO2, (e) (Ti0.9Zr0.1)O2, (f) (Ti0.9V0.1)O2, and (g) (Ti0.9Mn0.1)O2. A few reflections are due to XRD sample holder (aluminium). Combined (h) X-ray powder diffraction data of bare and doped TiO2 (vertical bars are reflection markers) and (i) variation of cell volume vs. ionic radii with a line as a guide to the eye. | ||
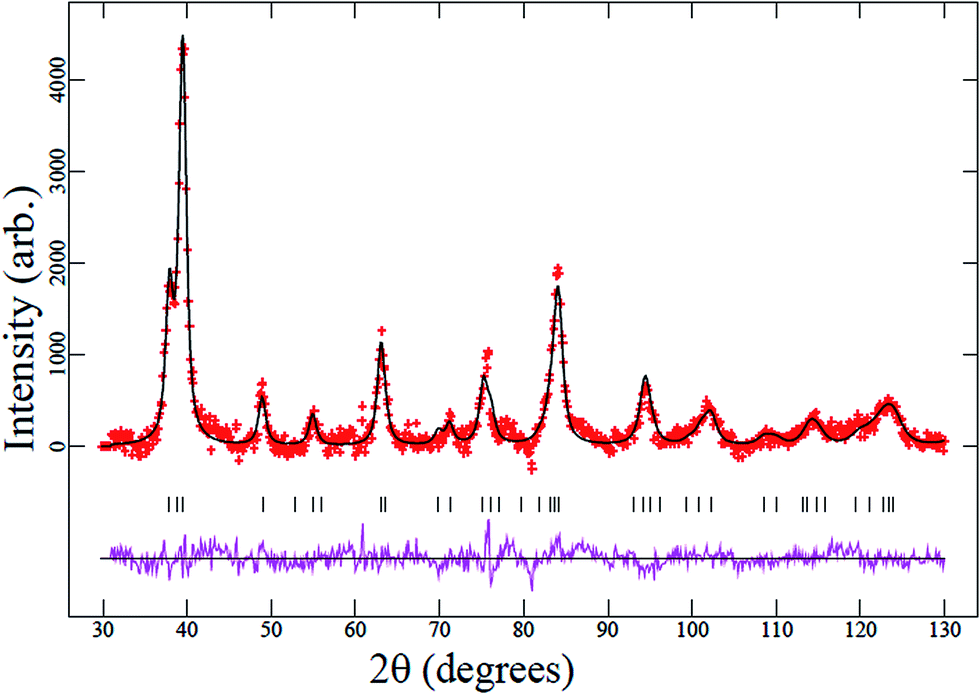 | ||
| Fig. 2 Rietveld-refinement profile using the anatase Ti0.9Mn0.1O2 model with NPD data. The red crosses are the data, the black line through the crosses is the calculated Rietveld model, the purple line below is the difference between the data and calculated model, and the vertical black-lines are reflection markers for anatase. | ||
| a (Å) | c (Å) | Assumed oxidation state of M′ or M′′ | Ionic radii (pm) | |
|---|---|---|---|---|
| TiO2 | 3.7890(1) | 9.4909(4) | Ti(IV) | 74.5 |
| (Ti0.9Fe0.1)O2−y | 3.8010(1) | 9.5035(4) | Fe(II) low spin | 75 |
| Fe(III) | 69 | |||
| Fe(IV) | 72.4 | |||
| (Ti0.9Zr0.1)O2 | 3.7982(1) | 9.5769(4) | Zr(IV) | 86 |
| (Ti0.9V0.1)O2 | 3.7841(1) | 9.4799(4) | V(V) | 68 |
| (Ti0.9Mn0.1)O2 | 3.7894(1) | 9.4533(4) | Mn(II) | 81 |
| Mn(III) | 72 | |||
| Mn(IV) | 67 | |||
| 0.1SnO2·0.9TiO2 | 3.7919(1) | 9.5258(4) | Sn(IV) | 83 |
| 0.1NiO·0.05Nb2O5·0.85TiO2 | 3.7941(1) | 9.4984(4) | Ni(II) | 83 |
| Nb(V) | 78 |
Data from Table 1 show that the variation of the unit-cell volume relative to pure TiO2, even for the composite materials, follows the expected trend based on the ionic radius of the dopant ion. This suggests that some cation substitution is occurring in the TiO2 component in the composite materials, i.e., a small proportion of the transition metals are substituting at the Ti site. A decrease in the unit-cell volume is observed for (Ti0.9V0.1)O2 and (Ti0.9Mn0.1)O2 which can be rationalised by the smaller ionic-radii of V5+ and Mn4+ relative to Ti4+ (Fig. 1i).32 This indicates that Mn is oxidised from the original oxidation-state of Mn2+ to Mn4+. (Ti0.9Zr0.1)O2 exhibits the largest change in lattice parameter to a = 3.7982(1) Å and c = 9.5769(4) Å, which corresponds to Zr4+ featuring the largest ionic-radii of the substituted samples. X-ray photo electron spectroscopy (XPS) data show the presence of Ti4+, Mn4+, Zr4+, Sn4+, V5+, and Fe3+ (Table 2). The fitted data of core level spectra of all compounds are shown in Fig. 3. In all compounds Ti2p shows two peaks at binding energies (BE) of ∼458 and ∼464 eV, and the latter peak is due to multiple splitting of the Ti-ion (Fig. 3, Table 2). The difference in binding energy (2p3/2 and 2p1/2 of Ti2p) ΔE of bare and doped TiO2 compounds shows a value of ∼5.7 eV, which is characteristic of TiO2 (Fig. 3). The Ni/Nb-doped sample shows BE of 206.83 and 209.53 eV, which corresponds to splitting of 3d5/2 and 3d3/2 found in Nb2O5 (ref. 33) for Nb, and Ni2p3/2 is observed at a BE of 855.4 eV corresponding to Ni2+.34 The multiple splitting of the energy levels of the Ni-ion gives rise to satellite peaks around 861.67 eV. The O1s main peak at a BE of 529 eV is found some samples which may in part be due to surface oxygen. Additional BE of other atoms in these samples are also shown in Fig. 3.
| Material | Binding energy Ti Ti2p3/2, Ti2p1/2 (eV) | Binding energy O1s (eV) | Binding energy of doped element (eV) | BET surface-area (±0.2 m2 g−1) |
|---|---|---|---|---|
| TiO2 | 458.09, 463.78 | 529.32 | — | 106 |
| 531.47 | ||||
| 533.07 | ||||
| Ti0.9Sn0.1O2 (0.1SnO2·0.9TiO2) | 458.38, 464.07 | 529.63 | Sn3d5/2: 486.18 | 135.7 |
| 531.13 | Sn3d3/2: 494.60 | |||
| Ti0.9Fe0.1O2 | 458.02, 463.72 | 529.25 | Fe2p3/2: 710.75 | 92.6 |
| 530.92 | Fe2p1/2: 723.32 | |||
| Ti0.9Zr0.1O2 | 458.35, 464.05 | 529.62 | Zr3d5/2: 181.65 | 158.8 |
| 531.20 | Zr3d3/2: 184.23 | |||
| Ti0.9Mn0.1O2 | 458.05, 463.74 | 529.28 | Mn2p3/2: 641.95 | 103 |
| 531.54 | Mn2p1/2: 653.52 | |||
| Ti0.9V0.1O2 | 458.14, 463.84 | 529.41 | V2p3/2: 516.73 | 74 |
| 531.27 | V2p1/2: 524.36 | |||
| 0.1NiO·0.05Nb2O5·0.85TiO2 | 458.54, 463.78 | 529.71 | Nb3d5/2: 206.83 | 104.3 |
| 531.74 | Nb3d3/2: 209.53 | |||
| Ni2p3/2: 858.40 | ||||
| Ni2p1/2: 861.67 |
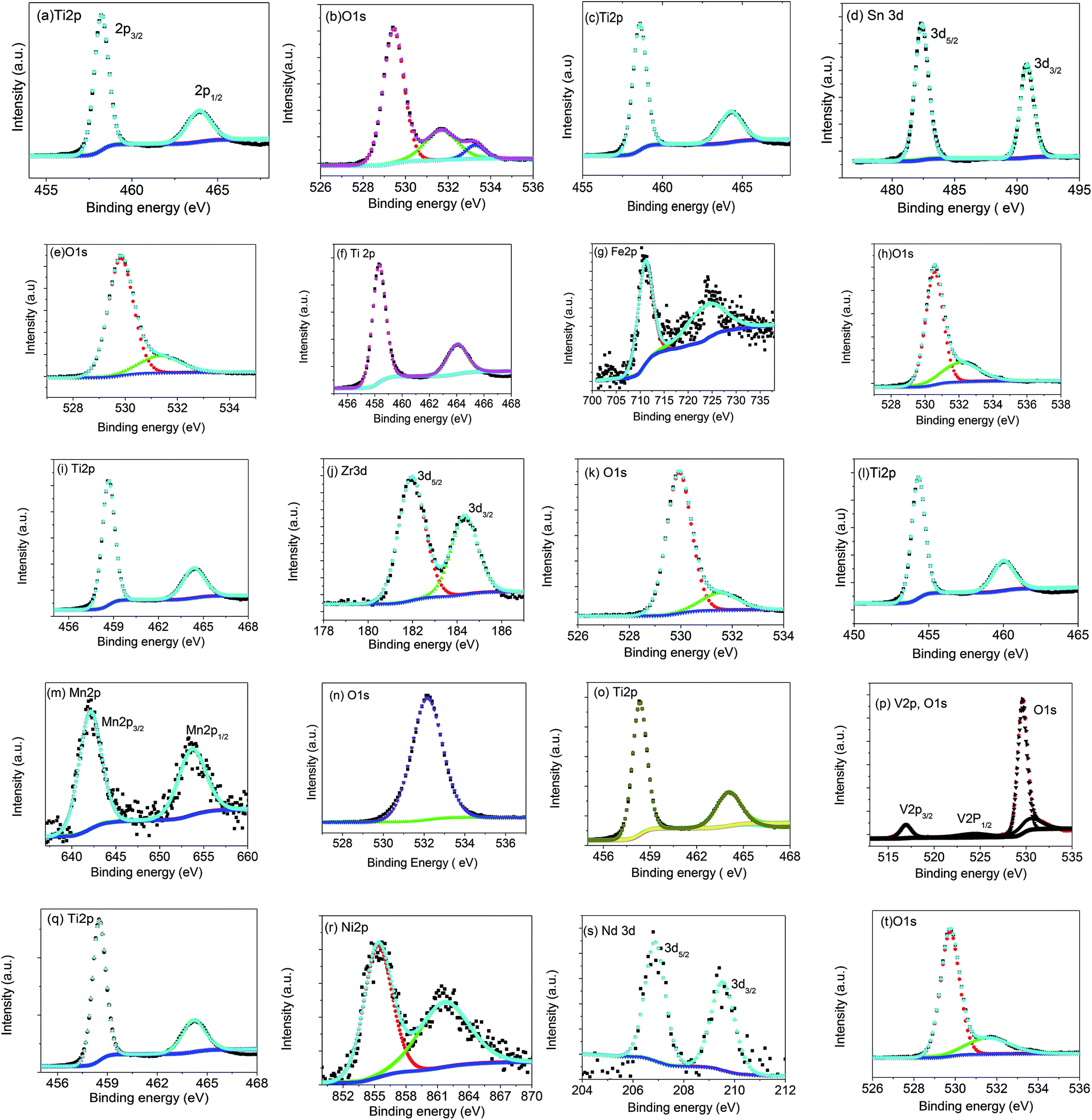 | ||
| Fig. 3 Fitted core level XPS spectra of bare and doped TiO2 (a and b) bare TiO2, (c–e) 0.1SnO2·0.9TiO2, (f–h) (Ti0.9Fe0.1)O2, (i–k) (Ti0.9Zr0.1)O2, (l–n) (Ti0.9Mn0.1)O2, (o and p) (Ti0.9V0.1)O2, (q–t) 0.1NiO·0.05Nb2O5·0.85TiO2. Experimental (black), fitted (light blue, red and green) and background corrections (blue) symbols are shown. | ||
NPD provided further details of the cation substitution and oxygen vacancies, due to the better sensitivity of neutrons towards near-neighbour elements in the periodic table, e.g. Mn and Ti, and lighter elements, e.g. O, in the presence of heavier ones, relative to X-rays. Manganese was substituted onto the titanium site in the ideal anatase model and the figures-of-merit for the Rietveld refinement were Rp = 2.20, wRp = 2.88, and χ2 = 1.68 for 17 refineable parameters. The fit to the NPD data is shown in Fig. 2. The oxygen positional-parameter refined to z = 0.1656(2) and the atomic displacement parameters (ADPs) of the mixed titanium/manganese and oxygen sites were found to be 0.0113(12) and 0.0166(6) Å2, respectively. No evidence was found for oxygen vacancies in accordance with the presumed oxidation state Mn4+ which also complements XPS results (Table 2). An approximate average crystallite-size was calculated from the peak-profile functions (particle-size broadening) and found to be ∼200 nm. To determine the Ti:Mn ratio the ADP of the Ti/Mn site was fixed and the Ti:Mn ratio was refined with the total occupancy constrained to 1. The ratio was found to be 0.90(17):0.10(17) in good agreement with the expected composition.
Raman scattering was used as a non-destructive probe of the lattice vibrational-modes of metal oxides in the synthesized materials. According to factor group analysis, the 15 normal modes of anatase TiO2 have the irreducible representation 1A1g + 1A2u + 2B1g + 1B2u + 3Eg + 2Eu. The A1g, B1g, and Eg modes are Raman active, and thus six fundamental-transitions are expected in the Raman spectrum of anatase.35Fig. 4 shows the four main Raman bands observed at 200, 396, 515, and 637 cm−1 in pure TiO2, which correspond to the typical Raman bands for anatase at room temperature found in the literature at 198, 398, 518, and 639 cm−1.36 The relative intensities of the transition-metal containing TiO2 are normalized for comparative purposes and also shown in Fig. 4. Raman spectra of the synthesized samples, whether transition-metal substituted or composite, exhibit overlapping peaks with TiO2 indicating that they have the same underlying Ti vibrational-modes as anatase TiO2. This is expected for the composite electrodes as the anatase component is relatively unchanged, and illustrates that the doping at this level the substituted electrodes has a minor influence on the anatase signal. The dominant fraction electrochemical performance can be related to titanium in a similar vibrational-state in pure, substituted, and composite electrodes.
 | ||
| Fig. 4 Raman spectra of transition-metal containing TiO2. Pure TiO2 shows Raman bands at 200, 396, 515, and 637 cm−1. | ||
The electrochemical activity of an anode can be influenced by the surface area available for reactions. Table 2 shows typical BET data for the synthesized materials. (Ti0.9Zr0.1)O2 shows the highest surface-area of 159 m2 g−1 and (Ti0.9V0.1)O2 shows the smallest of only 74 m2 g−1. The transition-metal substituted electrodes (Ti0.9Fe0.1)O2 and (Ti0.9Mn0.1)O2 as well as the composite 0.1NiO–0.05Nb2O5–0.85TiO2 have surface areas that are comparable to pure TiO2 (Table 2).
TEM was used to reveal the morphology of the synthesized samples and their selected-area electron diffraction (SAED) patterns are shown in Fig. 5. The average particle-size of the synthesized materials was ∼5–25 nm which is a direct consequence of the molten-salt synthesis method, with a significantly-lower synthetic temperature compared to other synthesis procedures such as conventional solid-state reactions. High-resolution TEM images show the (101) plane of anatase TiO2. The circular rings in the SAED patterns also indicate presence of anatase TiO2, consistent with XRD data, which seem to adopt nano-crystalline domains at this resolution.
 | ||
| Fig. 5 TEM images at different scales (including high resolution) and SAED images of pure TiO2 (a–c), 0.1SnO2·0.9TiO2 (d–f), (Ti0.9Fe0.1)O2 (g–i), 0.1NiO·0.05Nb2O5·0.85TiO2 (j–l), (Ti0.9Zr0.1)O2 (m–o), (Ti0.9Mn0.1)O2 (p–r), and (Ti0.9V0.1)O2 (s–u). | ||
The molten-salt method produces substituted compounds with transition metals M′ = Fe, Zr, Mn, and V, while others Sn and Nb/Ni result in composite compounds. Three of the compounds have a surface area comparable with pure TiO2 (M′ = Fe, Mn and M′′ = Nb/Ni). To determine the differences in anode performance of the synthesized compounds, charge–discharge cycling up to 70 cycles at a current density of 130 mA g−1 between 1.0 and 2.6 V was employed. Representative voltage–capacity profiles of the compounds are shown in Fig. 6. For clarity, only selected cycles are shown. The observed charge and discharge capacities are listed in Table 3. During discharge, lithium ions intercalate into TiO2, reducing Ti4+ to Ti3+ and resulting in a voltage plateau in the range 1.6–1.8 V, with the reverse occurring on charge resulting in a voltage plateau in the range 1.8–2.0 V. Li insertion/de-insertion during discharge–charge cycling for an isovalent dopant M can be represented as follows: TiO2 + zLi+ + ze− ↔ LizMx (Tiz3+Ti1−z4+)O2. The observed Li-poor37,38 and Li-rich37,38 regions are similar to previous reports.9,39 In the cases where M′ = V, Zr, Fe, and Mn (substituted electrodes) and to some extent in the Sn composite, a second plateau or at least an anomaly is observed in the charge curves. This may indicate some electrochemical influence of the transition-metal substitution on the charge–discharge curve. Interestingly, the Nb/Ni composite electrode shows similar charge-profiles to pure TiO2. The discharge profiles are fairly similar among this combination of transition-metal inclusions and resulting materials.
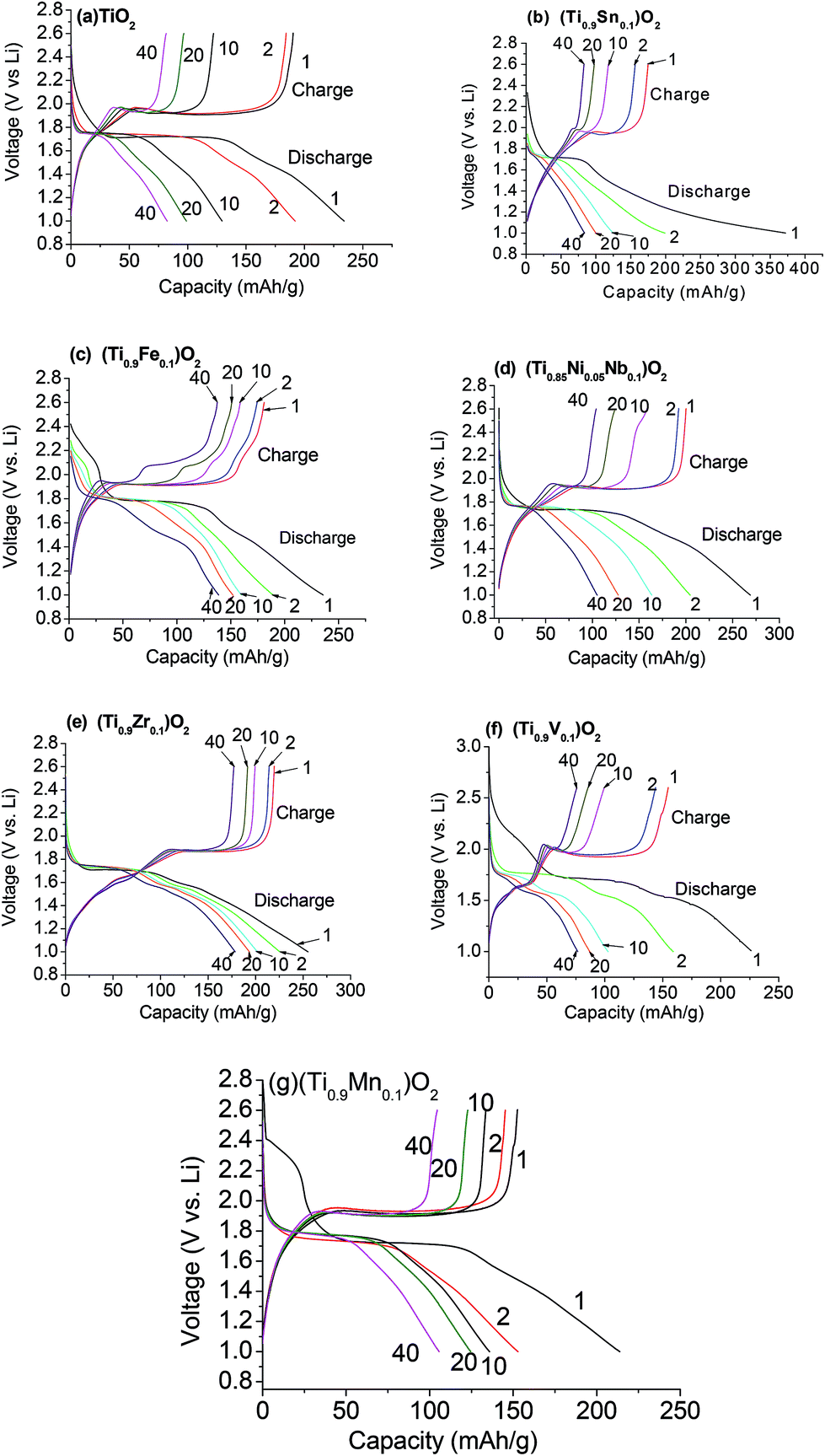 | ||
| Fig. 6 Voltage vs. capacity profiles for (a) TiO2, (b) 0.1SnO2·0.9TiO2, (c) (Ti0.9Fe0.1)O2, (d) 0.1NiO·0.05Nb2O5·0.85TiO2, (e) (Ti0.9Zr0.1)O2, (f) (Ti0.9V0.1)O2, and (g) Ti0.9Mn0.1O2. Cycle numbers are indicated. Cycling voltage range 1.0–2.6 V vs. Li, current density 130 mA g−1. | ||
| Compound | First cycle capacity (±3 mA h g−1) | 10th cycle capacity (±3 mA h g−1) | 60th cycle capacity (±3 mA h g−1) | ICL (±3 mA h g−1) | Capacity fade (%) 2nd–60th cycle | |||
|---|---|---|---|---|---|---|---|---|
| Charge | Discharge | Charge | Discharge | Charge | Discharge | Discharge | Charge | |
| TiO2 | 190 | 235 | 129 | 122 | 73 | 74 | 45 | 61 |
| (Ti0.9Fe0.1)O2 | 181 | 235 | 159 | 160 | 125 | 126 | 46 | 28 |
| (Ti0.9Zr0.1)O2 | 220 | 255 | 199 | 202 | 161 | 162 | 29 | 24 |
| (Ti0.9V0.1)O2 | 155 | 159 | 99 | 101 | 69 | 70 | 12 | 52 |
| (Ti0.9Mn0.1)O2 | 153 | 213 | 134 | 136 | 87 | 88 | 40 | 43 |
| 0.1SnO2·0.9TiO2 | 175 | 372 | 118 | 125 | 76 | 76 | 173 | 51 |
| 0.1NiO·0.05Nb2O5·0.85TiO2 | 200 | 269 | 158 | 163 | 91 | 92 | 64 | 53 |
The capacity vs. cycle number plots of the as-synthesized compounds are shown in Fig. 7 and selected values listed in Table 3. (Ti0.9Zr0.1)O2 has the least capacity fade, at about 24% and also a small irreversible capacity loss (ICL) of 29 mA h g−1. The Zr and Fe substituted and Ni/Nb composite samples show superior capacity-retention relative to the pure anatase TiO2, with the Ni/Nb composite having a capacity retention that is comparable with pure anatase TiO2 above approximately 30 cycles. Among all, the (Ti0.9Zr0.1)O2 compound delivers the best electrochemical performance of 160 mA h g−1 at the end of the 60th cycle, this maybe in part due to the larger surface area of this sample compared to the others. The V substituted and Sn composite samples show inferior performance to pure anatase TiO2. The possible cause for the ICL in these samples is likely to be a result of lithium being used for the formation of a solid-electrolyte interface (SEI) layer. This is perpetuated by the high surface-area of the as-synthesized compounds, especially during the first discharge.
 | ||
| Fig. 7 Capacity vs. cycle plots of pure and doped TiO2 from 1st to at least 60 cycles in the voltage range 1.0–2.6 V vs. Li with a current density of 130 mA g−1. Filled and open symbols represent discharge and charge capacities, respectively. | ||
Isovalent Zr substitution of Ti in anatase TiO2 shows the best performance after extended cycling (e.g. capacity retention and ICL), even though there is a small anomaly in the charge curves (Fig. 6). The lower-valent Fe substitution is comparable with the best-performing Zr-substituted sample with the exception of a higher ICL. The lower absolute capacity at the end of cycling will be influenced by the lower surface-area compared to the Zr-doped sample. The higher valent V-substitution shows the lowest ICL of 12 mA h g−1, presumably related to its smallest surface-area, although the capacity is inferior relative to pure TiO2. Arguably the worst performing sample was the Sn-containing composite. The Nb/Ni composite has a surface-area and charge–discharge profile similar to pure TiO2 and as shown in Fig. 6, 7 and Table 3 the extended cycling-behavior is similar to pure TiO2. The composite is predominantly TiO2, possibly with some Ni substitution at the Ti site, and a small amount of Nb/Ni-containing secondary phase, with these minor changes not significantly influencing the capacity of the electrode even though the overall Ti content has been reduced. We note that electrochemical properties are sensitive to preparation method, temperature and time which will influence morphology of TiO2 materials. Here we have kept the synthetic conditions identical, future work can now modify synthetic conditions, an additional parameter, of the samples with favourable performance to further optimise these electrode materials. Further studies on the bulk conductivity of undoped and doped TiO2 bulk pellets and also within a Li-ion cell, in addition to complementary spectroscopy techniques such as X-ray absorption spectroscopy may shed further light on the observed electrochemical behaviour.
The cyclic voltammograms (CV) of pure TiO2, (Ti0.9Fe0.1)O2, 0.1SnO2·0.9TiO2, 0.1NiO·0.05Nb2O5·0.85TiO2, Ti0.9Mn0.1O2, and Ti0.9V0.1O2 are shown in Fig. 8. The lower negative-current of the CV represents reduction of Ti4+ to Ti3+ and Li-ion intercalation, and the upper positive-part of the CV represents oxidation where Ti3+ is oxidized back to Ti4+ and Li-ion de-intercalation.40 For the intercalation process, the main peak is between 1.7 and 1.8 V and for the de-intercalation process the main peak is at ∼2.0 V. The features between the samples are quite similar with the exception of the Fe, Mn, Zr, and V-containing samples showing another positive feature presumably related to electrochemical activity of Fe, Mn, Zr, and V couples as postulated above for the differences in the charge curves. The overlapping nature of all 6 cycles indicates relatively-good reversibility of the charge–discharge reaction.
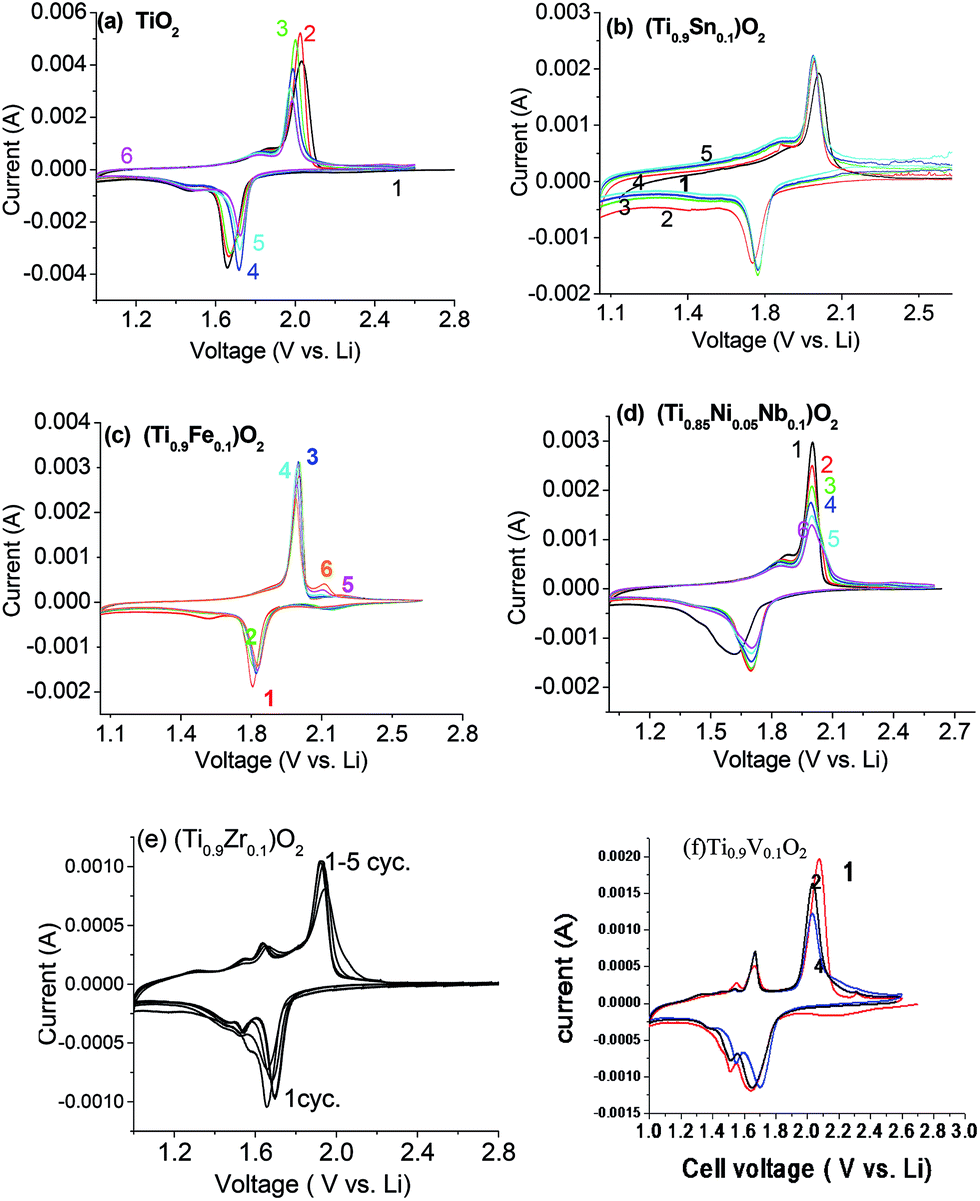 | ||
| Fig. 8 Cyclic voltammograms of (a) TiO2, (b) 0.1SnO2·0.9TiO2, (c) (Ti0.9Fe0.1)O2, (d) 0.1NiO·0.05Nb2O5·0.85TiO2, (e) Ti0.9Mn0.1O2, and (f) Ti0.9V0.1O2 at a scan rate of 0.058 mV s−1 at room temperature. The numbers refer to cycle numbers. | ||
4. Conclusion
To explore transition-metal substitution in TiO2 and composite TiO2 anodes, samples were synthesized using the molten-salt method at 410 °C. Rietveld analysis of XRD and NPD data, as well as XPS and Raman analysis revealed that the major component in all compounds is anatase. TEM results show that the synthesized particles are ∼5–25 nm. Transition metals M′ = Fe, Zr, Mn, and V result in a doped compound while M′ = Sn and Nb/Ni result in a composite compound. BET surface-area analysis show that (Ti0.9Zr0.1)O2 has the largest surface-area of ∼159 m2 g−1 while (Ti0.9V0.1)O2 has the smallest surface-area of ∼74 m2 g−1, with (Ti0.9Fe0.1)O2, (Ti0.9Mn0.1)O2, and 0.1NiO–0.05Nb2O5–0.85TiO2 having surface areas that are comparable with pure TiO2. Electrochemical studies, galvanostatic charge–discharge and cyclic voltammetry, show that (Ti0.9Zr0.1)O2 features the best battery-performance, of 160 mA h g−1 at the end of the 60th cycle, of any of the electrodes studied (including pure TiO2) with only ∼24% capacity fade between 2 to 60 cycles. The composite electrode 0.1SnO2–0.9TiO2 exhibits the highest irreversible capacity-loss of 173 mA h g−1 with capacity fade of 51%. Extra features in the charge and CV profiles of the M′ = V, Zr, Fe and Sn samples were observed which indicate the possibility of the electrochemical activity of another redox couple.Overall, performance is not necessarily improved by doping. The observed multiphase composite-anodes show no significant enhancement of capacity retention, while certain single-phase substitutions, likely depending on the oxidation state of the substituting cation, results in clear performance enhancements. Indications are that isovalent substitutions with the molten-salt method provide the best battery performance. This work shows that in order to systematically improve the electrochemical performance by homogeneous doping (direct cation-substitution) or heterogeneous doping (the formation of secondary phases), samples have to be synthesized under identical conditions. This has to be undertaken in conjunction with systematic determination of particle-size, extent of Mn, Zr doping in to TiO2, preparation time (generally synthetic parameters) and surface-area and the same electrochemical controls (comparing against samples with similar values for these parameters) in order to definitively state whether a doping regime does improve performance or whether there are other factors that contribute to the changes.
Acknowledgements
MVR, RPR, SA wish to thank National Research Foundation, Prime Minister's Office, Singapore under its Competitive Research Programme (CRP Award no. NRF-CRP 10-2012-6, NRF-CRP 8-2011-4) for research support. Support for this project was provided in part by the ANSTO funded “Energy Materials” research project within the Bragg Institute. Dr Sharma would like to thank AINSE Ltd for providing support through the research fellowship scheme. Authors thank to Mr Henche Kuan, Dept. of MSE, NUS for recording XPS data.References
- Z. G. Yang, D. Choi, S. Kerisit, K. M. Rosso, D. H. Wang, J. Zhang, G. Graff and J. Liu, J. Power Sources, 2009, 192, 588–598 CrossRef CAS.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- S. H. Nam, H. S. Shim, Y. S. Kim, M. A. Dar, J. G. Kim and W. B. Kim, ACS Appl. Mater. Interfaces, 2010, 2, 2046–2052 CAS.
- P. N. Zhu, Y. Z. Wu, M. V. Reddy, A. S. Nair, B. V. R. Chowdari and S. Ramakrishna, RSC Adv., 2012, 2, 531–537 RSC.
- D. Barreca, G. Carraro, A. Gasparotto, C. Maccato, M. Cruz-Yusta, J. L. Gomez-Camer, J. Morales, C. Sada and L. Sanchez, ACS Appl. Mater. Interfaces, 2012, 4, 3610–3619 CAS.
- W. Q. Lu, C. M. Lopez, N. Liu, J. T. Vaughey, A. Jansen and D. W. Dees, J. Electrochem. Soc., 2012, 159, A566–A570 CrossRef CAS PubMed.
- R. A. Leising, M. J. Palazzo, E. S. Takeuchi and K. J. Takeuchi, J. Electrochem. Soc., 2001, 148, A838–A844 CrossRef CAS PubMed.
- P. Strobel, F. Le Cras and M. Anne, J. Solid State Chem., 1996, 124, 83–94 CrossRef CAS.
- G. Sudant, E. Baudrin, D. Larcher and J. M. Tarascon, J. Mater. Chem., 2005, 15, 1263–1269 CAS.
- Y. L. Qi, Y. D. Huang, D. Z. Jia, S. J. Bao and Z. P. Guo, Electrochim. Acta, 2009, 54, 4772–4776 CrossRef CAS PubMed.
- G. D. Du, N. Sharma, V. K. Peterson, J. A. Kimpton, D. Z. Jia and Z. P. Guo, Adv. Funct. Mater., 2011, 21, 3990–3997 CrossRef CAS PubMed.
- Y. Z. Wu, M. V. Reddy, B. V. R. Chowdari and S. Ramakrishna, Electrochim. Acta, 2012, 67, 33–40 CrossRef CAS PubMed.
- Z. G. Yang, J. L. Zhang, M. C. W. Kintner-Meyer, X. C. Lu, D. W. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- M.-H. Ryu, K.-N. Jung, K.-H. Shin, K.-S. Han and S. Yoon, J. Phys. Chem. C, 2013, 117, 8092–8098 CAS.
- J. Qiu, S. Li, E. Gray, H. Liu, Q.-F. Gu, C. Sun, C. Lai, H. Zhao and S. Zhang, J. Phys. Chem. C, 2014, 118, 8824–8830 CAS.
- P. Afanasiev and C. Geantet, Coord. Chem. Rev., 1998, 178, 1725–1752 CrossRef.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, J. Power Sources, 2006, 160, 1369–1374 CrossRef CAS.
- M. V. Reddy, P. Zhu, Y. Wu, S. Peng, S. Yang, A. Sreekumaran Nair, K. P. Loh, B. V. R. Chowdari and S. Ramakrishna, Chem. Commun., 2012, 48, 10865–10867 RSC.
- M. V. Reddy, C. Yu, F. Jiahuan, K. P. Loh and B. V. R. Chowdari, RSC Adv., 2012, 2, 9619–9625 RSC.
- X. Zhao, M. V. Reddy, H. Liu, S. Ramakrishna, G. V. Subba Rao and B. V. R. Chowdari, RSC Adv., 2012, 2, 7462–7469 RSC.
- M. V. Reddy, Z. Beichen, L. J. Nicholette, Z. Kaimeng and B. V. R. Chowdari, Electrochem. Solid-State Lett., 2011, 14, A79–A82 CrossRef CAS PubMed.
- M. V. Reddy, K. Y. H. Kenrick, T. Y. Wei, G. Y. Chong, G. H. Leong and B. V. R. Chowdari, J. Electrochem. Soc., 2011, 158, A1423–A1430 CrossRef CAS PubMed.
- A. Sakunthala, M. V. Reddy, S. Selvasekarapandian, B. V. R. Chowdari and P. C. Selvin, Energy Environ. Sci., 2011, 4, 1712–1725 CAS.
- A. J. Studer, M. E. Hagen and T. J. Noakes, Phys. B, 2006, 385-386, 1013–1015 CrossRef CAS.
- N. Sharma, M. V. Reddy, G. D. Du, S. Adams, B. V. R. Chowdari, Z. P. Guo and V. K. Peterson, J. Phys. Chem. C, 2011, 115, 21473–21480 CAS.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- N. Sharma and V. K. Peterson, J. Solid State Electrochem., 2012, 16, 1849–1856 CrossRef CAS.
- R. P. Rao, N. Sharma, V. K. Peterson and S. Adams, Solid State Ionics, 2013, 230, 72–76 CrossRef CAS PubMed.
- M. V. Reddy, C. T. Cherian, K. Ramanathan, K. C. W. Jie, T. Y. W. Daryl, T. Y. Hao, S. Adams, K. P. Loh and B. V. R. Chowdari, Electrochim. Acta, 2014, 118, 75–80 CrossRef CAS PubMed.
- M. V. Reddy, C. Yu, F. Jiahuan, K. P. Loh and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2013, 5, 4361–4366 CAS.
- R. P. Rao, M. V. Reddy, S. Adams and B. V. R. Chowdari, Mater. Res. Bull., 2015, 66, 71–75 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- A. Le Viet, M. V. Reddy, R. Jose, B. V. R. Chowdari and S. Ramakrishna, J. Phys. Chem. C, 2010, 114, 664–671 CAS.
- K. S. Tan, M. V. Reddy, G. V. SubbaRao and B. V. R. Chowdari, J. Power Sources, 2005, 141, 129–142 CrossRef CAS PubMed.
- L. J. Hardwick, M. Holzapfel, P. Novák, L. Dupont and E. Baudrin, Electrochim. Acta, 2007, 52, 5357–5367 CrossRef CAS PubMed.
- R. Baddour-Hadjean, S. Bach, M. Smirnov and J. P. Pereira-Ramos, J. Raman Spectrosc., 2004, 35, 577–585 CrossRef CAS PubMed.
- M. Wagemaker, A. P. M. Kentgens and F. M. Mulder, Nature, 2002, 418, 397–399 CrossRef CAS.
- M. Wagemaker, W. J. H. Borghols and F. M. Mulder, J. Am. Chem. Soc., 2007, 129, 4323–4327 CrossRef CAS.
- M. V. Reddy, S. Adams, G. T. J. Liang, I. F. Mingze, H. Van Tu An and B. V. R. Chowdari, Solid State Ionics, 2014, 262, 120–123 CrossRef CAS PubMed.
- M. V. Reddy, X. W. V. Teoh, T. B. Nguyen, Y. Y. M. Lim and B. V. R. Chowdari, J. Electrochem. Soc., 2012, 159, A762–A769 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |