
Synthesis of cadmium oxide and carbon nanotube based nanocomposites and their use as a sensing interface for xanthine detection†
Abstract
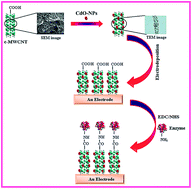
Xanthine oxidase (XOD) extracted from bovine milk was immobilized covalently via N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxy succinimide (NHS) chemistry onto cadmium oxide nanoparticles (CdO)/carboxylated multiwalled carbon nanotube (c-MWCNT) composite film electrodeposited on the surface of an Au electrode. The nanocomposite modified Au electrode was characterized by Fourier transform infrared (FTIR), cyclic voltammetry (CV), scanning electron microscopy (SEM) and electrochemical impedance spectroscopy (EIS) before and after immobilization of XOD. Under optimal operation conditions (25 °C, +0.2 V vs. Ag/AgCl, sodium phosphate buffer, pH 7.5), the following characteristics are attributed to the biosensor: linearity of response up to xanthine concentrations of 120 μM, detection limit of 0.05 μM (S/N = 3) and a response time of at most 4 s. After being used 100 times over a period of 120 days, only 50% loss of the initial activity of the biosensor was evaluated when stored at 4 °C. The fabricated biosensor was successfully employed for the determination of xanthine in fish meat.
 Please wait while we load your content...
Please wait while we load your content...

