
Cu(ClO4)2·6H2O catalyzed solvent free per-O-acetylation and sequential one-pot conversions of sugars to thioglycosides†
Debnath Chatterjee,
Abhijit Paul,
Rajkamal and
Somnath Yadav*
Department of Applied Chemistry, Indian School of Mines, Dhanbad-826004, Jharkhand, India. E-mail: yadav.s.ac@ismdhanbad.ac.in; Fax: +91-326-229-6563; Tel: +91-326-223-5880
First published on 20th March 2015
Abstract
The solvent free per-O-acetylation of various reducing and non-reducing sugars has been carried out using stoichiometric amounts of acetic anhydride and copper(II) perchlorate hexahydrate as the catalyst. The reactions with various reducing monosaccharides have also been followed by a one-pot sequential conversion to the corresponding thioglycosides in high yields.
Introduction
Per-O-acetylation is perhaps the most used primary protecting group reaction in carbohydrate synthesis. Its importance stems from the fact that the introduction of the acetyl protecting group can be carried out under a variety of conditions, the products are very stable under many reaction conditions and they can be used as glycosyl donors under Lewis acidic conditions. Again the acetate protecting group can be easily cleaved using catalytic amounts of NaOMe (Zemplen's method). They are also used routinely for their conversion to glycosyl halides and thioglycosides which have found widespread use as glycosyl donors, the latter particularly for iterative glycosylations.1 The classical method for carrying out the per-O-acetylation reaction uses pyridine as the solvent as well as the base. Sometimes, to accelerate the reactions DMAP is also added.2 However, several issues with this such as the use of a large excess of the toxic and foul smelling pyridine have led to the development of many improved catalytic methods. Among the various catalysts used for the conversion are (i) bases such as NaOAc,3 NaOH/TBAB,4 imidazole5 and DABCO6 (ii) protic acids such as H2SO4,7 H3BO3/H2SO4 (ref. 8) and p-toluene sulfonic acid9 (iii) Lewis acids such as ZnCl2,10 FeCl3,11 BF3–Et2O,12 Cu(OTf)2,13 Sc(OTf)3,14 In(OTf)3,15 Ce(OTf)3,16 LiClO4,17 and Fe2(SO4)3 (ref. 18) (iv) heterogeneous catalysts such as montmorillonite K-10,19 H-β-zeolite,20 Amberlyst-15,21 H2SO4–SiO2,22 HClO4–SiO2,23 molecular sieves,24 Al2O3,25 sulphonic acid fuctionalized γ-Al2O3 (ref. 26) and other catalysts such as I2 (ref. 27) and N-bromosuccinimide.28Although effective many of these methods suffer from a few limitations such as the use of large excess of acetic anhydride, use of volatile organic solvents (VOSs) and on many occasions the toxicity and high cost of the catalysts. Therefore effective catalysts that are economically more feasible, environmentally more sustainable and use stoichiometric amounts of acetic anhydride and avoids the use of VOSs are desirable. Towards this objective, we have explored a few cheap and easily available Cu2+ salts for carrying out the per-O-acetylation of free sugars and report that Cu(ClO4)2·6H2O is a very effective catalyst for carrying out the desired reaction under solvent free conditions at room temperature using only stoichiometric amounts of Ac2O. We have also followed up the solvent free, Cu(ClO4)2·6H2O catalyzed per-O-acetylation of free reducing monosaccharides using stoichiometric Ac2O with the sequential one pot thioglycosylation reaction using BF3–Et2O to generate the corresponding thioglycosides. The one-pot per-O-acetylation–thioglycosylation strategy for the synthesis of thiosugars has previously been employed with p-toluene sulfonic acid,9 Cu(OTf)2,13 I2,27 and SnCl4 (ref. 29) in conjunction with BF3–Et2O. However these methods suffer from the limitation that the catalysts are often very corrosive, have a very short shelf life and often are very costly.
Results and discussion
The preliminary experiments for the solvent free per-O-acetylation reaction was carried out using D-glucose (1a) and Ac2O with a few commercially available hydrated Cu(II) salts (Table 1). The experiments with 10 mol% of Cu(OAc)2·H2O, CuCl2·2H2O and Cu(NO3)2·3H2O using an excess of Ac2O under solvent free conditions yielded very poor results. In each case the reactions were very slow and less than 50% of the per-O-acetylated product 2a could be isolated after 6 days (entries 1–3, Table 1). The reaction using CuSO4·5H2O as catalyst at 10 mol% was slightly better but once again the reaction yielded only about 79% of 2a after 3 days (entry 4, Table 1). In contrast to all the above, the reaction with 10 mol% of Cu(ClO4)2·6H2O afforded 2a in quantitative yield after only 30 minutes (entry 5, Table 1). In an effort to lower the catalyst loading, the reaction was then carried out using only 5 mol% of Cu(ClO4)2·6H2O with only 5.05 equivalents of acetic anhydride when it was observed that the catalyst retained its activity without any decrease in the yield (entry 6, Table 1). In the final effort towards optimizing the conditions for the reaction, it was carried out using only a stoichiometric amount of Ac2O and 1 mol% of Cu(ClO4)2·6H2O when it was found that the yield was once again nearly quantitative (entry 7, Table 1).
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Ac2O equiv. | Time | Yieldb |
| a The reactions were carried out with 1 g of 1a in neat.b Isolated yields. | ||||
| 1 | Cu(OAc)2·H2O (10) | 7.5 | 6 days | 58 |
| 2 | CuCl2·2H2O (10) | 7.5 | 6 days | 46 |
| 3 | Cu(NO3)2·3H2O (10) | 7.5 | 6 days | 28 |
| 4 | CuSO4·5H2O (10) | 7.5 | 4 days | 79 |
| 5 | Cu(ClO4)2·6H2O (10) | 7.5 | 0.5 h | 97 |
| 6 | Cu(ClO4)2·6H2O (5) | 5.05 | 0.5 h | 95 |
| 7 | Cu(ClO4)2·6H2O (1) | 5.05 | 0.5 h | 97 |
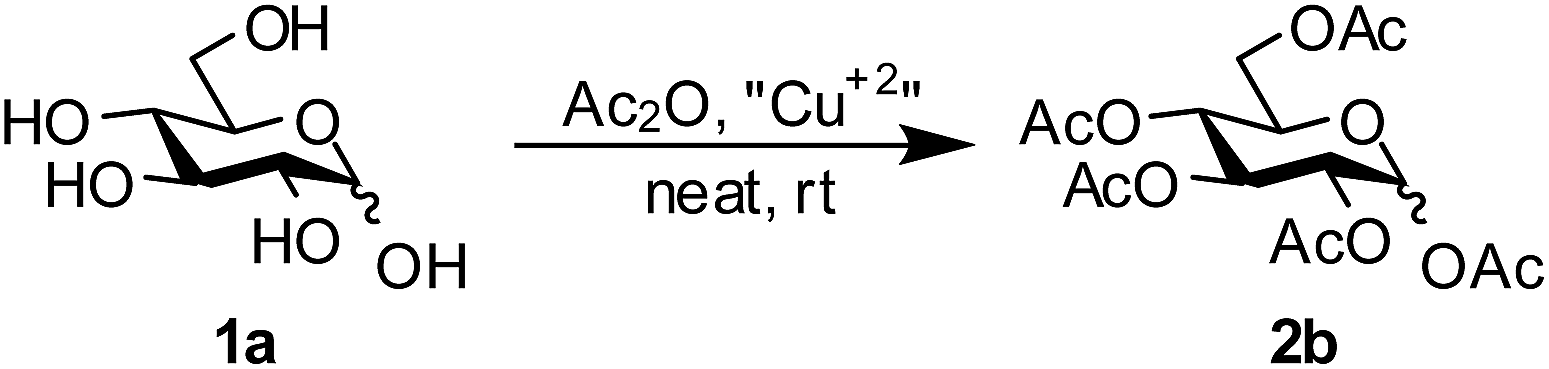
Following the initial success for the per-O-acetylation reaction using Cu(ClO4)2·6H2O, the optimized reaction conditions were employed for the per-O-acetylation of a series of unprotected sugars. The reactions were carried out under solvent free conditions using stoichiometric amounts of Ac2O and 1 mol% of Cu(ClO4)2·6H2O. The products of the per-O-acetylation reactions with various substrates are shown in Table 2. The per-O-acetylation of the corresponding fully unprotected reducing sugars such as mannose, galactose and xylose resulted in the formation of the fully acetylated products 2b (95%), 2c, (91%) and 2g, (90%). The reactions were very fast and were completed in only about 15 minutes. The procedure was also applied for the synthesis of several per-O-acetylated derivatives of non-reducing monosaccharides such as the 2,3,4,6-tetra-O-acetyl-α-methyl glucopyranoside (2h, Table 2), per-O-acetyl-myo-inositol (2i, Table 1) and per-O-acetyl-D-mannitol (2j, Table 2). Various reducing amino sugars and their derivatives such as N-acetylglucosamine, N-phthalimidoglucosamine and 2-deoxy-2-azidoglucose were also per-O-acetylated under the standard conditions in very good yields (2d–f, Table 2). However, the reactions with N-phthalimidoglucosamine and 2-deoxy-2-azidoglucose were slower and required 3 hours and 2.5 hours respectively for completion. The per-O-acetylation reaction was also successful with disaccharides such as maltose and sucrose leading to products 2k and 2l in almost quantitative yields as also the cyclic sugar β-cyclodextrin 2m. All the products were characterized by 1H and 13C spectrometry and the data corresponded well with the literature values.
| a Reagents and conditions for the per-O-acetylation reactions: Ac2O (1.01 equivalents per –OH), Cu(ClO4)2·6H2O (1 mol%), neat, rt. |
|---|
 |
The plausible mechanism of the reaction appears to be through the acylium perchlorate intermediate B (Scheme 1) formed by the reaction of Cu(ClO4)2 and Ac2O.31 Intermediate B would be able to acetylate the free alcohol moieties very efficiently. Again the efficiency of the copper perchlorate in the reaction is possibly due to the solubility of the former in acetic anhydride which may be ascribed to the reaction between the two, especially since the acylium perchlorate intermediate has been detected by NMR previously.31 The other Cu2+ salts are much less soluble in the reaction medium and probably are not able to form sufficiently long lived acylium intermediates to enable the acetylation reaction.
 | ||
| Scheme 1 Plausible mechanism of the per-O-acetylation reaction using Cu(ClO4)2·6H2O. | ||
After the success with the solvent free per-O-acetylation of the fully unprotected monosaccharides using stoichiometric amounts of acetic anhydride and Cu(ClO4)2·6H2O, we turned our attention towards the sequential one-pot thioglycosylation reaction with monosaccharides. Thioglycosides have come to be widely accepted and used as glycosyl donors due to their long shelf life, stability and ease of activation with easily available promoters.30 Again the thioglycosides are the most useful donors in iterative glycosylation strategies which enable the synthesis of oligosaccharides rapidly in one-pot.1 The thioglycosylation of anomeric acetates is traditionally carried out using excess of Lewis acids such as ZnCl2,32 BF3–Et2O33 or SnCl4.34 We employed the second one for our reaction and carried out the sequential one-pot per-O-acetylation–thioglycosylation for a series of fully unprotected sugars. The results are summarized in Table 3. A typical procedure involved treating the unprotected monosaccharide with stoichiometric amount of Ac2O in the presence of 1 mol% of Cu(ClO4)2·6H2O. After completion of the per-O-acetylation reaction, as evidenced by TLC, p-thiocresol was added to the reaction mixture followed by addition of 2 equivalents of BF3–Et2O. The reaction mixture was then allowed to stir at room temperature for the appropriate time till complete disappearance of the acetylated product.
|
|---|
| a Reagents and conditions: (i) Ac2O, Cu(ClO4)2·6H2O (1 mol%), neat, rt.; (ii) p-TolSH, BF3–Et2O (2 equivalents). |
 |
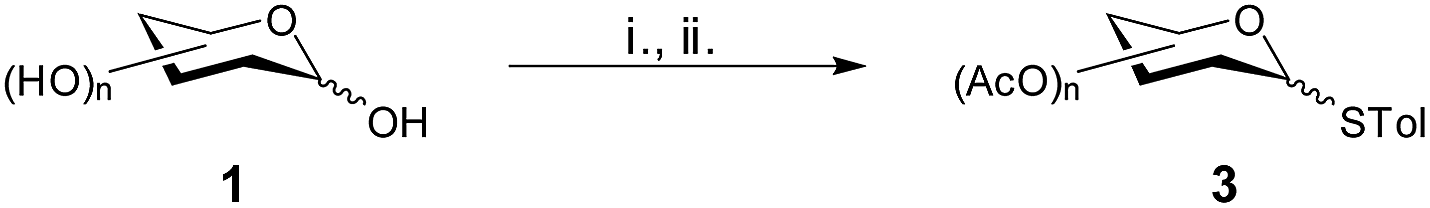
The sequential one-pot per-O-acetylation–thioglycosylation with glucose afforded the corresponding thioglycoside exclusively as the β isomer 3a in an overall yield of 70%. Mannose yielded the α-thioglycoside 3b in an overall yield of 82%. The reaction with galactose afforded the β-thiogalactopyranoside 3c exclusively in 75% yield. The one-pot peracetylation was also successful with various aminosugars. N-Acetylglucosamine exclusively yielded the per-O-acetylated β-N-acetylthioglycoside 3d in a yield of 55%. The reaction with 2-deoxy-2-phthalimidoglucose yielded the per-O-acetylated β-thioglycoside 3e exclusively in a yield of 70%, while 2-azido-2-deoxyglucose yielded a mixture of the per-O-acetylated α- and β-thioglycosides 3f in a ratio of 1.23/1 and an overall yield of 65%.
Experimental
General methods
All the starting sugars were either purchased from commercial sources or prepared according to the standard procedures. Anhydrous solvents were prepared using standard methods. TLC was performed on precoated aluminium plates of silica gel 60 F254. Flash column chromatography was carried out with silica gel 60 (230–400 mesh, E. Merck) at medium pressure. NMR spectra were recorded either on a 400 MHz or a 500 MHz spectrometer in solution of CDCl3 using tetramethylsilane as the internal standard, δ values are reported in parts per million (ppm) and coupling constants (J) in Hertz (Hz).General procedure for per-O-acetylation of carbohydrate substrates
To a mixture of sugar (1 g scale) and stoichiometric acetic anhydride (1.01 equivalents per –OH of sugar) was added Cu(ClO4)2·6H2O (1 mol% of sugar) under nitrogen and the mixture was kept stirring at room temperature. When TLC showed complete conversion of the starting material, the reaction mixture was diluted with ethyl acetate and the mixture was washed with aqueous NaHCO3 followed by brine. The ethyl acetate extract was then dried over anhydrous Na2SO4, the mixture was filtered, and the filtrate was concentrated in vacuo. The crude product was purified by flash column chromatography to afford the expected compound.Compound characterization data
General procedure for one-pot per-O-acetylation–thioglycosylation of sugars
Per-O-acetylation of sugar was carried out as described above. When reaction was completed according to TLC, p-thiocresol (2 equiv.) and BF3–Et2O (2 equiv.) were sequentially added to the reaction solution, and the mixture was allowed to stir for 2 days. The reaction was quenched by addition of aqueous NaHCO3 and the mixture was extracted with ethyl acetate three times. The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. Purification of the residue through flash column chromatography gave the desired thioglycoside.Compound characterization data
Conclusions
In summary, we have reported a very convenient method of the per-O-acetylation of unprotected sugars using the very cheap and easily available Cu(ClO4)2·6H2O. The per-O-acetylation reactions could be carried out under solvent free conditions using a stoichiometric amount of acetic anhydride. The reactions required very less time and used a very low loading of only 1 mol% of the Cu(ClO4)2·6H2O catalyst. Furthermore the Cu(ClO4)2·6H2O catalyzed per-O-acetylation reaction was followed by a sequential thioglycosylation reaction to afford the corresponding per-O-acetylated thioglycosides in good yields.Acknowledgements
We thank the Science and Engineering Research Board, GOI for financial assistance through Grant no. SR/FT/CS-130/2011. We also thank Sophisticated Analytical Instrument Facility, Punjab University and the NMR facility at University of Kalyani for the NMR analysis.Notes and references
- For some reviews on the use of thioglycoside donors in iterative glycosylation see: (a) C.-Y. I. Liu, S. Mulani and K.-K. T. Mong, Adv. Synth. Catal., 2012, 354, 3299 CrossRef CAS; (b) J. D. C. Codee, R. E. J. N. Litjens, L. J. van den Bos, H. S. Overkleeft and G. A. van der Marel, Chem. Soc. Rev., 2005, 34, 769 RSC; (c) X. Huang, L. Huang, H. Wang and X.-S. Ye, Angew. Chem., Int. Ed., 2004, 43, 5221 CrossRef CAS PubMed.
- G. Höfle, W. Steglich and H. Vorbrüggen, Angew. Chem., Int. Ed., 1978, 17, 569 CrossRef.
- M. L. Wolfrom and A. Thompson, Methods Carbohydr. Chem., 1963, 211 CAS.
- W. Szeja, Pol. J. Chem., 1980, 54, 1301 CAS.
- P. Tiwari, R. Kumar, P. R. Maulik and A. K. Misra, Eur. J. Org. Chem., 2005, 4265 CrossRef CAS.
- R. Ch, M. Tyagi, P. R. Patil and K. P. R. Kartha, Tetrahedron Lett., 2011, 52, 5841 CrossRef CAS PubMed.
- J. A. Hyatt and G. W. Tindall, Heterocycles, 1993, 35, 227 CrossRef CAS PubMed.
- R. H. Furneaux, P. M. Rendle and I. M. Sims, J. Chem. Soc., Perkin Trans. 1, 2000, 2011 RSC.
- C.-S. Chao, M.-C. Chen, S.-C. Lin and K.-K. T. Mong, Carbohydr. Res., 2008, 343, 957 CrossRef CAS PubMed.
- C. Limousin, J. Cleophax, A. Petit, A. Loupy and G. Lukacs, J. Carbohydr. Chem., 1997, 16, 327 CrossRef CAS.
- F. Dasgupta, P. P. Singh and H. C. Srivastava, Carbohydr. Res., 1980, 80, 346 CrossRef CAS.
- G. Agnihotri, P. Tiwari and A. K. Misra, Carbohydr. Res., 2005, 340, 1393 CrossRef CAS PubMed.
- C.-A. Tai, S. S. Kulkarni and S.-C. Hung, J. Org. Chem., 2003, 68, 8719 CrossRef CAS PubMed.
- J. C. Lee, C.-A. Tai and S.-C. Hung, Tetrahedron Lett., 2002, 43, 851 CrossRef CAS.
- N. P. Bizier, S. R. Atkins, L. C. Helland, S. F. Colvin, J. R. Twitchell and M. J. Cloninger, Carbohydr. Res., 2008, 343, 1814 CrossRef CAS PubMed.
- G. Bartoli, R. Dalpozzo, A. D. Nino, L. Maiuolo, M. Nardi, A. Procopio and A. Tagarelli, Green Chem., 2004, 6, 191 RSC.
- K.-C. Lu, S.-Y. Hsieh, L. N. Patkar, C.-T. Chen and C.-C. Lin, Tetrahedron, 2004, 60, 8967 CrossRef CAS PubMed.
- L. Shi, G. Zhang and F. Pan, Tetrahedron, 2008, 64, 2572 CrossRef CAS PubMed.
- P. M. Bhaskar and D. Loganathan, Tetrahedron Lett., 1998, 39, 2215 CrossRef CAS.
- P. M. Bhaskar and D. Loganathan, Synlett, 1999, 129 CrossRef CAS PubMed.
- G. Fan, C. Liao, T. Fang, S. Luo and G. Song, Carbohydr. Polym., 2014, 112, 203 CrossRef CAS PubMed.
- J. Zhang, B. Zhang, J. Zhou, J. Li, C. Shi, T. Huang, Z. Wang and J. Tang, J. Carbohydr. Chem., 2011, 30, 165 CrossRef CAS.
- A. K. Misra, P. Tiwari and S. K. Madhusudan, Carbohydr. Res., 2005, 340, 325 CrossRef CAS PubMed.
- L. Cai, C. Rufty and M. Liquois, Asian J. Chem., 2014, 26, 4367 CAS.
- P. Tiwari and A. K. Misra, Carbohydr. Res., 2006, 341, 339 CrossRef CAS PubMed.
- L. Wu and Z. Yin, Carbohydr. Res., 2013, 365, 14 CrossRef CAS PubMed.
- (a) B. Mukhopadhyay, K. P. R. Kartha, D. A. Russel and R. A. Fields, J. Org. Chem., 2004, 69, 7758 CrossRef CAS PubMed; (b) K. P. R. Kartha and R. A. Field, Tetrahedron, 1997, 53, 11753 CrossRef CAS.
- X.-F. Sun, R.-C. Sun, L. Zhao and J.-X. Sun, J. Appl. Polym. Sci., 2004, 92, 53 CrossRef CAS.
- S. Yan, N. Ding, W. Zhang, P. Wang, Y. Li and M. Li, J. Carbohydr. Chem., 2008, 31, 571 CrossRef.
- W. Zhong and G. J. Boons, in Handbook of Glycosylation, ed. A. V. Demchenko, Wiley-VCH verlag GmbH and Co. KGaA, Weinheim, 2008 Search PubMed.
- R. Dalpozzo, G. Bartolli, L. Sambri and P. Melchiorre, Chem. Rev., 2010, 110, 3501 CrossRef CAS PubMed.
- R. U. Lemieux, Can. J. Chem., 1951, 29, 1079 CrossRef CAS.
- R. Ferrier and R. Furneaux, Methods Carbohydr. Chem., 1980, 8, 251 CAS.
- R. U. Lemieux, Can. J. Chem., 1955, 33, 109 CrossRef CAS PubMed.
- J. Vesely, M. Ledvina, J. Jindrich, D. Saman and T. Trnka, Collect. Czech. Chem. Commun., 2003, 68, 1264 CrossRef CAS.
- R. Mukherjee and E. M. Axt, Phytochemistry, 1984, 23, 2682 CrossRef CAS.
- H. Wu, Y. Shen, L.-Y. Fan, Y. Wan and D.-Q. Shi, Tetrahedron, 2006, 62, 7995 CrossRef CAS PubMed.
- U. S. Chowdhury, Tetrahedron, 1996, 52, 12775 CrossRef CAS.
- G. Ngoje and Z. Li, Org. Biomol. Chem., 2013, 11, 1879 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C-NMR spectra are available. See DOI: 10.1039/c5ra03461b |
| This journal is © The Royal Society of Chemistry 2015 |