
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Repair efficiency of clustered abasic sites by APE1 in nucleosome core particles is sequence and position dependent†
Vandana
Singh
,
Bhavini
Kumari
and
Prolay
Das
*
Department of Chemistry, Indian Institute of Technology Patna, Patna-800013, Bihar, India. E-mail: prolay@iitp.ac.in; Fax: +91 612 225 7383; Tel: +91 612 255 2057
First published on 26th February 2015
Abstract
Closely located multiple abasic sites or clustered abasic sites are highly mutagenic and potentially cytotoxic. They have been found to be repair resistant in several in vitro studies. We studied the efficiency of the repair of clustered abasic sites by the APE1 enzyme in nucleosome core particles (NCPs). Sequences having genomic importance as the core sequence of TATA box and CpG islands were used to assemble the NCPs where the abasic clusters are located around the A/T or G/C rich 0.5 positioning site of the NCPs. The thermodynamics of the binding and repair of the A/T or G/C encased clustered abasic sites in the NCPs by APE1 enzyme are reported herein for the first time that was monitored by Isothermal Titration Calorimetry (ITC). The A/T encased clustered abasic sites in the NCP showed greater binding affinity with APE1 than the G/C counterpart. A/T encased abasic sites are also cleaved faster to generate double strand breaks by APE1 enzyme as compared to the CpG island sequence in the NCP, albeit at much slower rate than the linear model. Although, the overall reactivity of the abasic sites is appreciably reduced in the NCPs, distinct differences exist in the processing of the abasic sites that are flanked by A/T or G/C rich sequence. Our study suggests that both sequence effect and nucleosomal positioning are important determinants for the repair efficiency of clustered abasic sites in NCPs.
1. Introduction
One of the most common and highly mutagenic types of DNA damage results from the removal of bases from DNA, creating apuridinic or apyrimidinic residues, commonly known as abasic site.1 In addition to the endogenously induced ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 abasic sites per cell per day, abasic sites are also created exogenously and as intermediates during repair of few other DNA damage lesions.2,3 Abasic sites are repaired mostly by the base excision repair (BER) pathway where the enzyme apurinic/apyrimidinic endonuclease (APE1) plays a pivotal role.4–6 A complicated situation is encountered when two or more abasic sites are present in close proximity giving rise to abasic clusters.7,8 Clustered abasic sites are frequently induced by ionizing radiation, chemicals and various other factors.9–11 The optimal activity of the APE1 enzyme on an abasic site in a cluster has been found to be compromised due to conformational constraints imposed by the presence of additional abasic sites in the vicinity.12–14 Clustered abasic lesions have been found to be repair refractive in linear, condensed and genomic DNA.15–17 Formation of double strand breaks (DSBs) has been found to be severely hindered during BER of bistranded clustered oxidative lesions in nucleosomes.18 Surprisingly, nonenzymatic generation of DSBs from the cleavage of clustered abasic sites in Nucleosome Core Particles (NCPs) are greatly accelerated compared to naked DNA,19 which refutes the concept of repair resistivity of abasic clusters as revealed by other researchers.
000 abasic sites per cell per day, abasic sites are also created exogenously and as intermediates during repair of few other DNA damage lesions.2,3 Abasic sites are repaired mostly by the base excision repair (BER) pathway where the enzyme apurinic/apyrimidinic endonuclease (APE1) plays a pivotal role.4–6 A complicated situation is encountered when two or more abasic sites are present in close proximity giving rise to abasic clusters.7,8 Clustered abasic sites are frequently induced by ionizing radiation, chemicals and various other factors.9–11 The optimal activity of the APE1 enzyme on an abasic site in a cluster has been found to be compromised due to conformational constraints imposed by the presence of additional abasic sites in the vicinity.12–14 Clustered abasic lesions have been found to be repair refractive in linear, condensed and genomic DNA.15–17 Formation of double strand breaks (DSBs) has been found to be severely hindered during BER of bistranded clustered oxidative lesions in nucleosomes.18 Surprisingly, nonenzymatic generation of DSBs from the cleavage of clustered abasic sites in Nucleosome Core Particles (NCPs) are greatly accelerated compared to naked DNA,19 which refutes the concept of repair resistivity of abasic clusters as revealed by other researchers.
NCPs are the fundamental building unit of the chromatin structure.20,21 They are formed when the two units of each of the four histone proteins, namely H2A, H2B, H3 and H4 are wrapped around by 1.65 tight helical turns of ∼146–147 base pairs of DNA.22 The DNA compaction brought about by this DNA packaging in the NCP and hence the morphology monitors the accessibility of the DNA to different enzymes including those required for the DNA repair.23 The efficiency of BER for the repair of Uracil (U), Thymine glycol (Tg) as well as abasic site lesions has been found to be modulated in NCPs by the location of the specific damage residues at unique superhelical positions. Repair of U residues by Uracil DNA Glycosylase (UDG) have been found to be much slower in NCPs as compared to the naked DNA irrespective of nucleosomal translational position.24 Moreover, the activity of UDG is modulated in NCPs in the presence of histone depending on the rotational orientation of the Us.25 The repair of Tg by NTH1 was found to be hindered when the lesion was projecting inwards and close to the nucleosomal dyad.26 However, photochemically induced clustered abasic sites in NCPs have been found to create DNA–histone crosslinks at superhelical 1.5 location leading to faster strand scission.19 The non-enzymatic accelerated cleavage of clustered abasic sites in NCP is attributed to the formation of Schiff's base between the aldehyde form of the abasic site and the amines of the lysine rich histone tail in proximity to the sharp kinking of the DNA sequence at the 1.5 superhelical position. As far as enzymatic processing of abasic sites is concerned, generalization of this observation of faster cleavage of abasic sites in NCPs could be an over simplification, since clustered lesions have been shown to be repair refractive in many systems. Nonetheless, the fate of clustered abasic sites in NCPs in the presence of the lesion specific enzyme APE1 is largely unknown. Moreover, it remains to be seen whether cleavage efficiency of abasic sites in NCP is a function of nucleosomal positioning only. With respect to the repair of abasic clusters in NCPs, it can be said that quantitative evaluations of the sequence dependence and the positioning effect on the cleavage efficiency have not been comprehensively determined.
Herein, we report the efficiency of processing of clustered abasic sites in NCPs by radiolabel free gel electrophoresis assays. Most importantly, for the first time, quantitative evaluations of thermodynamic parameters of the sequence effect on the repair of clustered abasic sites in NCPs are presented here through Isothermal Titration Calorimetry (ITC) studies. Our choice of the sequence and precise location of the clustered abasic sites gives an opportunity to study the APE1 induced repair of abasic sites in the less researched 0.5 superhelical location in NCPs. The DNA sequence chosen for the study represents sequences of genomic importance as in the TATA box core sequence and CpG island sequence that encase the abasic sites with either A/T or G/C rich bases respectively.27 G/C rich positioning sequence at 0.5 site has an inherent distortion that assists in nucleosomal positioning and hence influence nucleosomal stability. Our studies show that the DNA sequence has an utmost role to play not only in the processing of the clustered abasic sites, but also in nucleosomal positioning that directs the repair process.
2. Experimental section
2.1. Material and methods
HPLC purified synthetic oligonucleotides were obtained from Sigma Aldrich custom oligo service. T4 DNA ligase, APE1 and UDG enzyme was purchased from New England Biolabs (NEB), USA that uses recombinant sources and purified from E. coli. Urea, proteinase K, tris buffer, formamide, glycerol, CRT0044876, lead acetate, chemicals for buffer were purchased from Sigma Aldrich. SYBR Gold® nucleic acid stain was purchased from Invitrogen. Polyacrylamide gel electrophoresis (PAGE) images were acquired using a UVP Gel Doc-It 310 gel documentation system. Quantitative estimation in the gel image was done using Vision Work Ls Image acquisition and analysis software from UVP (UK).2.2. Generation of abasic sites
The clustered abasic sites were generated using 5 units of UDG enzyme at 37 °C for 1 h on linear A1A2–T1T2–B1B2 and A1A2–G1G2–B1B2 duplexes. To confirm the complete formation of abasic sites, the UDG treated samples were incubated with 0.1 M NaOH and visualized on 20% denaturing PAGE.2.3. Construction of DNA sequences
The DNA sequences used in this study are shown in Table 1. A1 and A2 are complementary with four base overhangs on one side. Similarly, B1 and B2 are complementary with four base overhang on one side. There are three separate cassettes of double stranded DNA (dsDNA) in which the two terminal dsDNA cassettes (A1A2 and B1B2) are end labeled at 5′ termini with tetramethyl rhodamine (TAMRA) dye and 6-carboxy fluorescein (FAM) respectively. The cassette of the middle sequence is variable. One of the cassettes has a TATA box mimicking sequence (T1T2) and the other has CpG island sequences (G1G2). The two terminal cassettes along with any one of the middle cassettes potentially give rise to two 147 bp long dsDNA upon double ligation, (A1A2–T1T2–B1B2 and A1A2–G1G2–B1B2) (Fig. 1). Initially, the concerned six sequences are heated at 90 °C for 5 min in the presence of 10 mM sodium phosphate buffer and subsequently brought to room temperature for annealing. Ligation of DNA sequences was done using 400 units of T4 DNA ligase in the presence of 10 mM MgCl2, 10 mM ATP, 1% PEG and 1× T4 DNA ligase buffer. Ligation was done overnight at 16 °C where total volume was kept constant at 20 μl, followed by an additional one-hour incubation at 37 °C. The double ligated product was extracted from 12% native PAGE using extraction buffer consisting of 10 mM magnesium acetate, 0.5 M ammonium acetate, 1 mM EDTA, and 0.01% (w/v) SDS. In this extraction buffer, DNA sample was kept overnight at 37 °C, vortexed, ethanol precipitated and redissolved in appropriate buffer solution for further downstream studies.| A1 | 5′-TAMRA CGG TGA GGT CGG TGT TAG TGC CTG TAA CTC GGT GTT AGA GCC TGT-3′ |
| A2 | 5′-PO42− AGT TAC AGG CTC TAA CAC CGA GTT ACA GGC ACT AAC ACC GAC CTC ACC G-3′ |
| B1 | 5′-PO42− AGA GCC TGT AAC TCG GTG TTA GAG CCT GTA ACT CGG TGT TAG AGC CTG TAA-3′ |
| B2 | 5′-FAM TTA CAG GCT CTA ACA CCG AGT TAC AGG CTC TAA CAC CGA GTT ACA GG-3′ |
| T1 | 5′-PO42− AAC TCG GTG TTA GAG CCT GTA ACA CTC TAU ATA AAA CGC ACT CTC GGT GTT-3′ |
| T2 | 5′-PO42− CTC TAA CAC CGA GAG TGC GTU TTA TAT AGA GTG TTA CAG GCT CTA ACA CCG-3′ |
| G1 | 5′-PO42− AAC TCG GTG TTA GAG CCT GTA ACA CTC CGU GCG GAG AAT ACT CTC GGT GTT-3′ |
| G2 | 5′-PO42− CTC TAA CAC CGA GAG TAT TCU CCG CAC GGA GTG TTA CAG GCT CTA ACA CCG-3′ |
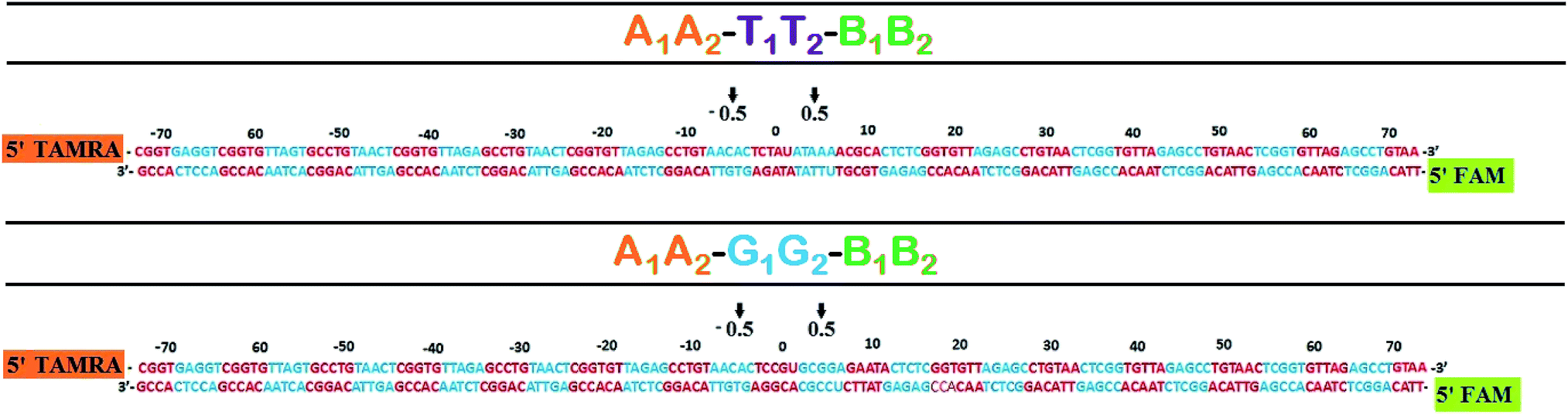 | ||
| Fig. 1 Histone–DNA registor of NCP constructs. The central base is numbered as 0 position and –ve and +ve signs are assigned for left and right respectively. 0.5 position indicates the probable superhelix location (SHL), turns from center. | ||
2.4. Reconstitution of NCP
EpiMark® Nucleosome Assembly Kit from NEB was used to construct NCP from 147 bp DNA. NCPs were reconstituted using dilution assembly protocol of NEB that yields 25 pmol of nucleosome giving maximum yield in the ratio of 1:1 octamer to DNA using 5 M NaCl. The concentration of NaCl was linearly reduced to 0.1 M using dilution buffer constituting 10 mM Tris, pH 8.0. The successful formation of NCP and the yield was analyzed on 8% (59:1 acrylamide:bisacrylamide) native PAGE in 1× TTE buffer. For downstream experiments, NCPs were selectively purified from double ligated product by dialysis against 0.10 M NaCl at 4 °C for 8 h with Hoefer CA membrane (MWCO, 100 kDa).
2.5. Denaturing and native PAGE
Denaturing PAGE (29:1 acrylamide–bisacrylamide cross linking ratio) in the presence of 7 M urea at pH 8.0 (1× TTE buffer) were run at a constant voltage of 200 V for 3 h to analyze percentage cleavage of abasic sites from DNA bands in the gel for linear model. 8% native PAGE (59:1 acrylamide–bisacrylamide cross linking ratio) was performed for the analysis of NCPs and to detect DSBs. 8% denaturing urea containing PAGE (59:1 acrylamide–bisacrylamide cross linking ratio) was performed to analyze percentage abasic sites cleavage in linear and NCP model. For inhibition studies, the DNA samples were incubated with 250 μM CRT0044876 and 20 μM lead acetate separately in the presence of two units of APE1 enzyme at 37 °C for 1 h before PAGE analysis. The experiments are done in triplicate for error minimization. For all the analysis, error bars were generated by calculation of the standard deviation from three data points. Unpaired t-tests were performed to monitor the statistical significance of the data.
2.6. Calculation of cleavage of abasic sites
The quantitative evaluation of percentage cleavage of abasic sites in A1A2–T1T2–B1B2 and A1A2–G1G2–B1B2 duplex was done from true color digital gel images as well as SYBR gold® stained gel images from CCD camera. A1–T1–B1 and A1–G1–B1 has abasic site (derived from Uracil) at the 75th position from the 5′-end. The corresponding complementary strands A2–T2–B2 and A2–G2–B2 have abasic site at 70th position from the 5′-end. Since there are two abasic sites located in opposite strands, maximum of four fragments are formed following complete cleavage by APE1.After reaction with APE1, A1–T1–B1 and A1–G1–B1 form two strand of 74 bases and 73 bases from the 5′ end respectively. Similarly A2–T2–B2 and A2–G2–B2 form two strands of 78 and 69 bases respectively. The amount of abasic sites cleaved is evaluated with the help of eqn (1).
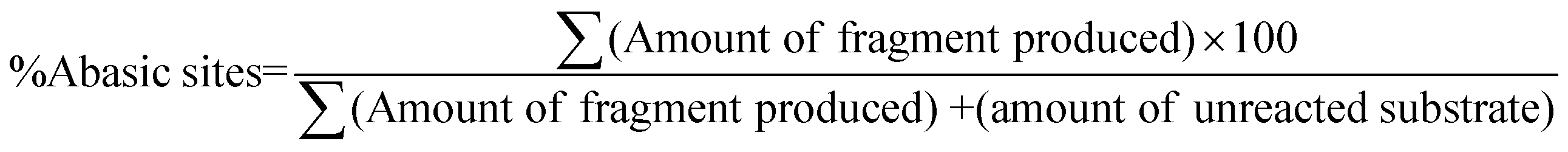 | (1) |
In native gels, each of the abasic sites containing 147 bp duplexes A1A2–T1T2–B1B2 and A1A2–G1G2–B1B2 yields two duplexes following APE1 treatment. For the estimation of DSBs from native PAGE, the following eqn (2) was used.
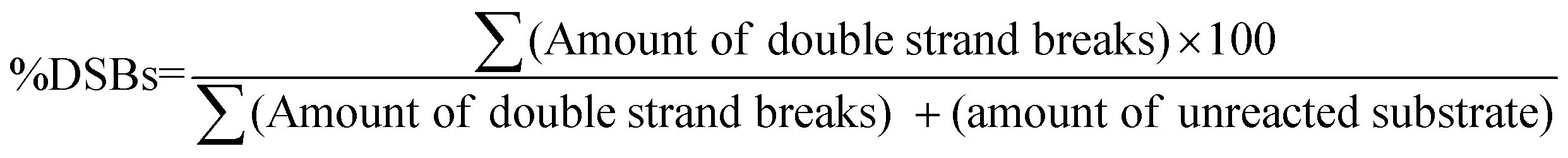 | (2) |
2.7. ITC measurement
ITC studies were conducted at 37 °C at pH 7.0 using MicroCal ITC200 (GE, USA). To determine the binding of the APE1 enzyme with the substrate, 0.8 μM of the 147 bp linear and NCP DNA were titrated against 0.08 μM of APE1 enzyme. Protein samples were dialyzed against 10 mM APE1 buffer (pH 7.0, devoid of Mg2+) for consistency in results. To further determine the binding of the APE1 enzyme with the substrate in the presence of the inhibitor, same concentration of enzyme and substrate was used. Buffer from the dialysis was used to resolubilize the DNA and to provide a non erroneous heat of dilution. Eqn (3) has been used to calculate the thermodynamic parameters.| ΔG = −RTlnKd = ΔH0 − TΔS | (3) |
3. Results and discussion
DNA sequence context is known to have a great effect on the nucleosomal positioning. The localized central 22 bases of 147 mer oligonucleotides have a profound effect on the crystal-packing configuration of nucleosomes.28 In addition to this, the repair of abasic sites in isolation or in clusters is highly influenced by the sequence context of the concerned DNA.27 This prompted us to investigate the processing of clustered abasic sites in the core sequence of TATA box and CpG island in the NCP model. The fact that the A/T or G/C sequences encase the clustered abasic sites that are located at critical positions in 147 bp oligomer duplex with respect to nucleosomal positioning, it offers a diligent opportunity to evaluate the effect of sequence and nucleosomal positioning on APE1 mediated repair of the clustered abasic sites.3.1. The yield of NCP is better with G/C rich cassette
The linear 147 bp DNA A1A2–T1T2–B1B2 and A1A2–G1G2–B1B2 are used to reconstitute NCPs. Better positioned nucleosome are formed with the sequence containing the core sequence of GC box i.e., A1A2–G1G2–B1B2. The yield of NCPs from the linear counterpart is higher by ∼20% for A1A2–G1G2–B1B2 as compared to A1A2–T1T2–B1B2 when the sequences are used as substrates for histones proteins (Fig. 2). In the present scenario, two 147 bp DNA with a difference of 12 base pair in the middle are reconstituted to NCP. Thus, the difference in nucleosomal positioning between A1A2–T1T2–B1B2 and A1A2–G1G2–B1B2 is solely due to the disparity of the 12 base pairs, where the former is A/T rich (TATA box mimicking sequence) and the latter is G/C rich (CpG island sequence) at the 0.5 position from the nucleosomal dyad. | ||
| Fig. 2 Reconstitution efficiency of A1A2–T1T2–B1B2 and A1A2–G1G2–B1B2 with histone proteins (A) 8% native PAGE stained with SYBR® gold. Lane 1 – ladder, lane 2 – A1A2–G1G2–B1B2 NCP, lane 3 – A1A2–T1T2–B1B2 NCP, lane 4 – control 147 mer, lane 5 – reaction products of double ligation (B) true color digital image of 8% native PAGE gel showing yield of NCPs. Lane numbering is same as (A). (C) Quantification and comparison of the yield of NCP reconstitution, where bar 1 – A1A2–G1G2–B1B2 NCP and bar 2 – A1A2–T1T2–B1B2 NCP. Each value represents mean ± SD and the *P < 0.0001. | ||
Gene regulation is coordinated via the nucleosomal organization.29 The binding affinity of histone octamer towards DNA sequence is highly variable and is clearly reflected in the distribution of nucleosomes in vivo.30,31 Multiple factors govern the organization of the NCP, the major being the DNA sequence context of the nucleosome along with the chromatin remodeler and site specific DNA-binding proteins.28 Reportedly, the GC/AT richness is the most important factor, which governs the nucleosome formation potential or inhibition respectively.32 GC rich sequences have been shown to display stronger nucleosome positioning due to their inherent flexibility, in contrast to AT rich sequences which have context dependent flexibility.33 Nucleosome inhibition has been observed with A tracts followed by a dinucleotide step.34 Thus the better yield of NCP for A1A2–G1G2–B1B2 observed here is a result of stronger nucleosomal positioning due to the presence of G/C rich sequences close to the nucleosomal dyad in the most probable position of +0.5. The presence of multiple abasic sites within this variable sequence poses an additional interesting question regarding their repair process, whether the repair process is histone imposed or sequence imposed.
3.2. Binding of APE1 with A/T encased abasic sites is more efficient
For the first time, we have used isothermal titration calorimetry to report thermodynamic parameters of the binding of APE1 enzyme with clustered abasic sites in NCPs (Fig. 3). The binding of APE1 enzyme with specific DNA molecule is an enthalpy driven process that contributes to the negative value of ΔG (Gibbs free energy) indicating the spontaneity of the process. We observed through ITC that the extent of spontaneity and binding of the enzyme with the abasic sites vary significantly in the two NCPs. Lowest negative enthalpy was obtained for the A1A2–G1G2–B1B2 NCP sequence (Table 2). Subsequently, the −ΔG value is lower for the A1A2–G1G2–B1B2 NCP compared to A1A2–T1T2–B1B2 NCP. This clearly shows that the binding of APE1 with the abasic sites is less efficient in the former. However, the value of the ΔG obtained through ITC experiments is less than that obtained by Adhikari et al., who studied the thermodynamic parameters of the binding of APE1 enzyme with single abasic residue by surface plasmon resonance experiments where the ΔG was estimated as ∼50 kJ mol−1.35 This is expected since the spontaneity of the enzyme binding decreases in the clustered abasic sites due to their poor processing. The lowest binding constant for A1A2–G1G2–B1B2 NCP is due to the inaccessibility of the binding sites to the APE1 enzyme. All the experiments were performed in the absence of Mg2+ ion, which is required for the cleavage of the abasic site following binding by the enzyme. This accounts for the low values of the binding constant, which represents solely the binding component. One noteworthy point is that in both the sequences, binding of the enzyme to the substrate is less in the NCPs compared to the linear counterpart. The substantial reduction in the APE1 enzyme activity in the NCPs may also be due to the preoccupancy of sites in the substrate by histone proteins.36 In the presence of specific APE1 inhibitors like CRT0044876 and Pb2+ the binding of the enzyme with the substrate do not decrease significantly. Interestingly, the same trend of higher binding affinity of A1A2–T1T2–B1B2 compared to A1A2–G1G2–B1B2 was observed in both linear and NCP samples. This shows that the binding preference of the enzyme to different DNA substrates in the presence of small molecule inhibitors like Pb2+ and CRT0044876 do not get affected much, it is the catalytic activity of the enzyme which is compromised. Summarily, the clustered abasic sites in the A/T rich TATA box mimicking sequence show better binding with APE1 enzyme in both the linear and the NCP model. Alternatively, it can be said that NCP of G/C rich CpG island sequence is more repair resistant in terms of clustered abasic damage and hence the lesions therein are longer lived. Thus, such complex lesions are susceptible to mutations and other mutagenic consequences.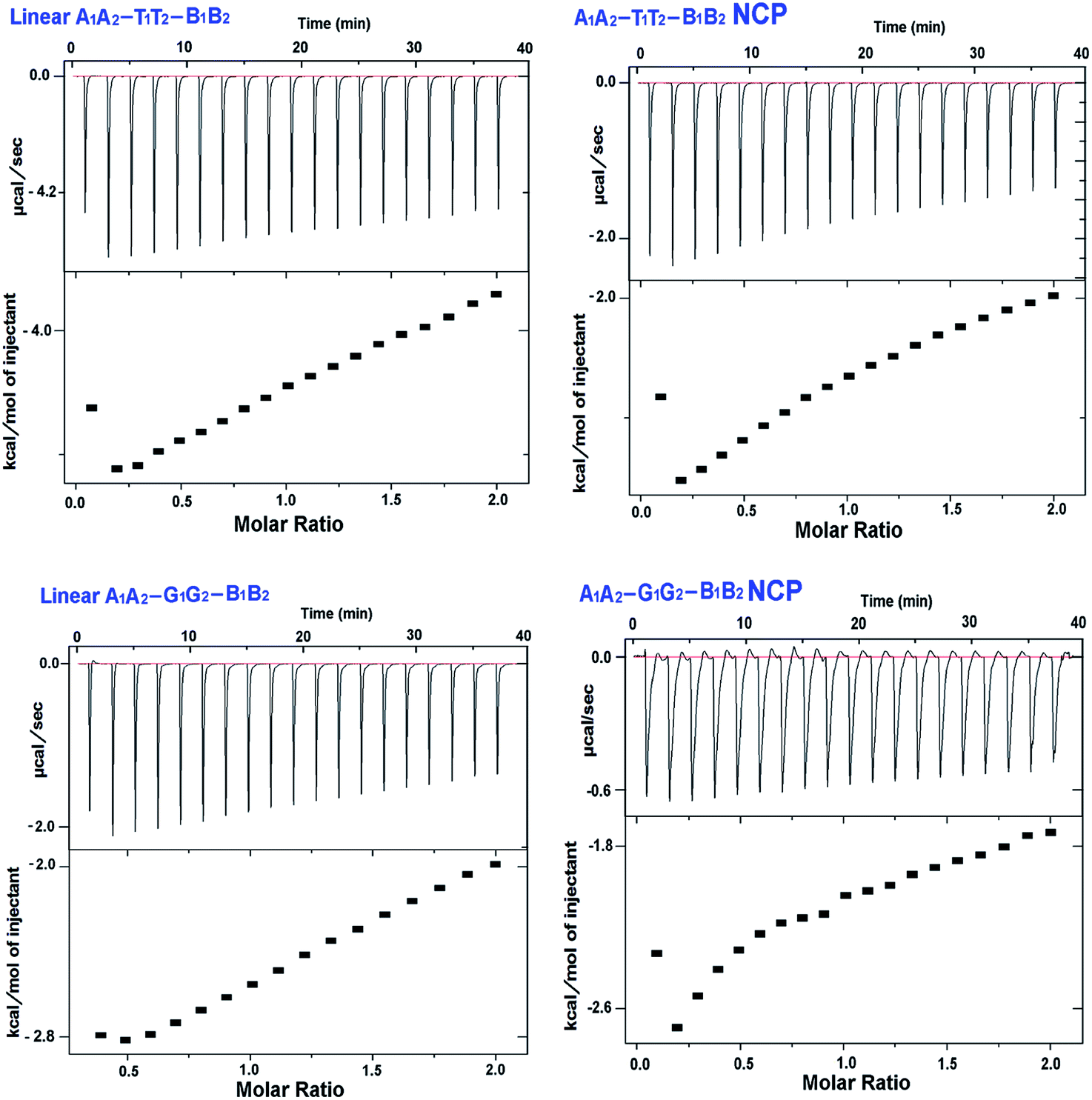 | ||
| Fig. 3 Isothermal titration calorimetry data for binding of APE1 enzyme to various DNA substrates containing clustered abasic sites. In each of the subfigures, the upper panel denotes the ITC titration curve and lower panel denotes binding isotherm of the action of APE1 enzyme. (A) A1A2–T1T2–B1B2 (B) A1A2–T1T2–B1B2 NCP (C) A1A2–G1G2–B1B2 (D) A1A2–G1G2–B1B2 NCP. | ||
| Sl. no. | Name of sample | K (M−1) | ΔH (kcal mol−1) | TΔS (kcal mol−1 K−1) | ΔG (kcal mol−1) |
|---|---|---|---|---|---|
| 1 | A1A2–T1T2–B1B2 | 2.31 × 104 | −5.03 ± 0.3 | 0.951 ± 0.3 | −5.99 ± 0.2 |
| 2 | A1A2–T1T2–B1B2 NCP | 4.09 × 103 | −4.38 ± 0.5 | 0.740 ± 0.2 | −5.12 ± 0.5 |
| 3 | A1A2–G1G2–B1B2 | 4.19 × 103 | −3.69 ± 0.2 | 1.450 ± 0.2 | −5.14 ± 0.2 |
| 4 | A1A2–G1G2–B1B2 NCP | 1.95 × 103 | −3.36 ± 0.1 | 1.174 ± 0.4 | −4.53 ± 0.1 |
3.3. Inefficient cleavage of G/C encased abasic sites in NCP contributes to repair refractivity
The sequences containing clustered abasic sites are cleaved with APE1 enzyme generating smaller fragments at the cleavage sites of the DNA. Herein, we have used FAM and TAMRA fluorescent probes to detect the fragments or the cleavage products in non-radiolabeled gel electrophoresis experiments. Parallely, we also performed gel analysis following SYBR® Gold staining and results were found to be similar.For the linear substrate A1A2–T1T2–B1B2, nearly ∼12 times higher substrate specificity for the APE1 enzyme was observed than A1A2–G1G2–B1B2. For A1A2–T1T2–B1B2, ∼4 times lower Km (Michaelis Menten constant) and ∼3 times higher Vmax,app than the A1A2–G1G2–B1B2 signifies that the catalytic efficiency of the A1A2–T1T2–B1B2 substrate is much higher than its GC rich counterpart (ESI, Fig. S5 and Table ST2†). Previously reported kinetic studies of APE1 enzyme with short oligomers (less than 50 bp) have demonstrated the dependence of the enzyme activity on the sequence in the proximity of the abasic residue as well as on the opposite bases in the complementary strand. This fact reinstates the importance of opposite and proximal sequence of the concerned abasic sites regarding the activity of APE1 enzyme. We found out that by using FAM and TAMRA labelled oligonucleotides, it is possible to directly assess which strand of the oligomer gets cleaved more by APE1 enzyme. In our case, we observed that the abasic sites surrounded by TTA TAT AGA and CCG CAC GGA are cleaved more which are tagged with FAM as compared to their complementary strand that was tagged with TAMRA (ESI Fig. S3 and S4†). The presence of CGG trinucleotide after five bases away from the abasic site might be a better substrate for APE1 as CGG has very low bending moduli, than that of B DNA.37 In case of the A/T rich sequence, the presence of AAA tract opposite to the FAM tagged strand might be assisting the cleavage of the abasic sites.38
The number of abasic sites cleaved in a given time interval was less in both the NCPs as compared to the corresponding linear 147 bp DNA. Among the NCPs, the amount of abasic sites cleaved in A1A2–G1G2–B1B2 NCP is ∼2.2 times less than that of A1A2–T1T2–B1B2 NCP (Fig. 4). This implies that in the linear DNA as well as in the NCPs, G/C richness around the abasic site is a dominating factor for lesser action of APE1 enzyme. Our results indicate limited processing of the G/C encased clustered abasic sites resulting from the restricted entry of the enzyme in the NCPs. The catalytic activity of APE1 is significantly reduced in the presence of inhibitors like CRT0044876 and Pb2+ (Fig. S1 and S2, ESI†). However, the trend of greater activity towards A/T encased abasic clusters is maintained by APE1, even in the presence of the inhibitors. This observation shows that excess of small molecule inhibitors is their in the NCPs, but do not contribute considerably in the sequence dependent reactivity of abasic sites towards APE1.
 | ||
| Fig. 4 Quantitative estimation of abasic sites in NCPs and linear model (A) true color denaturing PAGE image, Lane 1 and 2 – A1A2–G1G2–B1B2, lane 3 and 4 – A1A2–G1G2–B1B2 NCP, lane 5 and 6 – A1A2–T1T2–B1B2, lane 7 and 8 – A1A2–T1T2–B1B2 NCP. Samples of lane 2, 4, 6 and 8 are treated with 2 units of APE1 enzyme (B) denaturing PAGE stained with SYBR® gold where lane numbering are same as (A). Fragments with close number of bases appear to merge as single bands. (C) Quantification and comparison of abasic sites in linear and NCP models. Bar numbering are same as gel lanes. Each value represents mean ± SD and the *P < 0.0001 by two tailed t-test. | ||
Quantitative estimation of DSBs in NCPs after 10 min of reaction with APE1 enzyme shows that the number of DSBs created is ∼2.5 times more in A1A2–T1T2–B1B2 NCP than A1A2–G1G2–B1B2 NCP (Fig. 5). However, the DSBs formed in NCPs are much less in amount as compared to the DSBs formed in the corresponding linear model. The repair refractivity of the clustered abasic sites in the NCPs is further emphasized by this fact. Here, it is worth mentioning that not all the abasic sites formed are converted into potentially lethal DSBs. This is significant, since the system avoids formation of potentially lethal DSBs by this process. Understandably, clustered abasic sites in the NCP of CpG island sequences are processed much slower than the core sequence of TATA box and try to avoid formation of deleterious DSBs during the repair process. It is imperative that the sequence as well as packaging of the DNA determines the action of the APE1 enzyme. The significance of this result is that the deleterious clustered abasic sites become more repair refractive when reconstituted to nucleosomes. This effect is similar to the highly impaired APE1 mediated cleavage of clustered abasic sites in DNA condensates due to the restricted entry of the enzyme inside the condensates.16
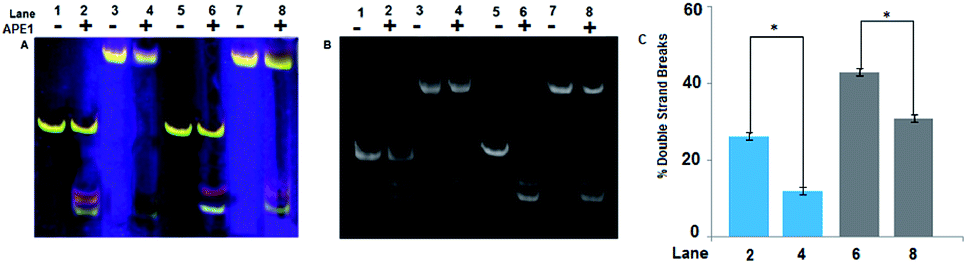 | ||
| Fig. 5 Quantitative estimation of DSBs in NCPs and linear model. (A) True color native PAGE image, lane 1 and 2 – A1A2–G1G2–B1B2, lane 3 and 4 – A1A2–G1G2–B1B2 NCP, lane 5 and 6 – A1A2–T1T2–B1B2, lane 7 and 8 – A1A2–T1T2–B1B2 NCP. Samples of lane 2, 4, 6 and 8 are treated with 2 units of APE1 enzyme and proteinase k. (B) Native PAGE stained with SYBR® gold where lane numbering are same as (A). (C) Quantification and comparison of DSBs in linear and NCP models. Bar numbering are same as gel lanes. Each value represents mean ± SD and the *P < 0.0001 by two tailed t-test. | ||
However, the repair refractivity of the abasic sites near the nucleosomal dyad as demonstrated here is different to what was reported by Greenberg et al., where they found DNA histone cross linking leading to faster strand scission at the 1.5 positioning site, although non-enzymatically.39 It is important to understand the difference in the location of the abasic sites in the concerned NCPs. As observed by them, abasic sites located in the 1.5 position with respect to the nucleosomal dyad are prone to faster processing, since this position is considered as a hot spot for DNA damage and binding activities. Considering the above and our findings along with that of Cannan et al.,18 the importance of nucleosomal positioning in dictating the efficiency of processing of DNA damage lesions has to be acknowledged. In our case, we didn't find any DNA–histone crosslinks, since their formation modifies gel mobility of DNA bands in PAGE, which was not observed. This also shows the importance of nucleosomal positioning in dictating the efficiency of processing of DNA damage lesions.
4. Conclusions
Our study regarding the processing of clustered abasic sites in NCPs reveal two important aspects of the repair process. Firstly, the efficiency of the cleavage of abasic sites was markedly reduced in NCPs compared to the naked DNA. Secondly, the cleavage of abasic sites located within G/C rich sequence close to the nucleosomal dyad in NCPs was even slower than those encased by A/T rich sequence. Evidence of slow and selective binding of the APE1 enzyme with the clustered abasic sites is provided herein by ITC studies. Lower binding affinity of APE1 with abasic sites in NCPs than the linear counterpart and comparatively better binding with A/T encased abasic sites was observed. Further confirmation of impaired APE1 activity in NCPs than naked DNA was obtained from gel electrophoresis experiments. Better cleavage efficiency of A/T encased abasic sites was also observed here. Nucleosomal positioning is believed to play an important role which directs the selective accessibility of the enzyme to the damaged lesions depending on the DNA sequence at the particular superhelical location. The slow processing of the abasic sites by APE1 in NCPs point towards the inherent repair refractivity of the lesions in clusters which is augmented by the restricted entry of the APE1 in the NCPs. This signifies that the abasic clusters are long lived in NCPs and hence prone to mutation. However, the slow processing ensures avoidance of the formation of potentially lethal DSBs that could lead to deleterious consequences.Acknowledgements
Funding from Department of Science and Technology (DST), Govt. of India (project no. SERB/LS/FT36/2010) to P. D is gratefully acknowledged. V. S. and B. K. are thankful to Central Scientific and Industrial Research (CSIR), Govt. of India (Sanction no. 09/1023(0004)/2010-EMR-1) and DST respectively for fellowship. IIT Patna is acknowledged for providing infrastructural and experimental facilities.Notes and references
- R. P. Cunningham, H. Ahern, D. Xing, M. M. Thayer and J. A. Tainer, Ann. N. Y. Acad. Sci., 1994, 726, 215–222 CrossRef CAS PubMed.
- M. M. Greenberg, Org. Biomol. Chem., 2007, 5, 18–30 CAS.
- T. Lindahl, Nature, 1993, 362, 709–715 CrossRef CAS PubMed.
- B. Demple and J. S. Sung, DNA Repair, 2005, 4, 1442–1449 CrossRef CAS PubMed.
- C. D. Mol, T. Izumi, S. Mitra and J. A. Tainer, Nature, 2000, 403, 451–456 CrossRef CAS PubMed.
- Y. Masuda, R. A. Bennett and B. Demple, J. Biol. Chem., 1998, 273, 30360–30365 CrossRef CAS PubMed.
- J. A. Mckenzie and P. R. Strauss, Biochemistry, 2001, 40, 13254–13261 CrossRef CAS PubMed.
- A. G. Georgakilas, Mol. BioSyst., 2008, 4, 30–35 RSC.
- B. M. Sutherland, P. V. Bennett, O. Sidorkina and J. Laval, Biochemistry, 2000, 39, 8026–8031 CrossRef CAS PubMed.
- J. Nakamura, V. E. Walker, P. B. Upton, S. Y. Chiang, Y. W. Kow and J. A. Swenberg, Cancer Res., 1998, 58, 222–225 CAS.
- P. Das, P. V. Bennet and B. M. Sutherland, J. Biosci., 2011, 36, 105–116 CrossRef CAS.
- B. Paap, D. M. Wilson III and B. M. Sutherland, Nucleic Acids Res., 2008, 36, 2717–2727 CrossRef CAS PubMed.
- S. Cunniffe, A. Walker, R. Stabler, P. O'Neill and M. E. Lomax, Int. J. Radiat. Biol., 2014, 90, 468–479 CrossRef CAS PubMed.
- M. A. Chaudhry and M. Weinfeld, J. Biol. Chem., 1997, 272, 15650–15655 CrossRef CAS PubMed.
- S. Bellon, N. Shikazono, S. Cunniffe, M. Lomax and P. O'Neill, Nucleic Acids Res., 2009, 37, 4430–4440 CrossRef CAS PubMed.
- V. Singh and P. Das, DNA Repair, 2013, 12, 450–457 CrossRef CAS PubMed.
- B. M. Sutherland, P. V. Bennett, O. Sidorkina and J. Laval, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 103–108 CrossRef CAS.
- W. J. Cannon, B. P. Tsang, S. S. Wallace and D. S. Pederson, J. Biol. Chem., 2014, 289, 19881–19893 CrossRef PubMed.
- J. T. Sczepanski, R. S. Wonga, J. N. McKnight, G. D. Bowman and M. M. Greenberg, Proc. Natl. Acad. Sci. U. S. A., 2010, 52, 22475–22480 CrossRef PubMed.
- S. Chakravarthy, Y. Park, J. Chodaparambil, R. S. Edayathumangalam and K. Luger, FEBS Lett., 2005, 579, 895–898 CrossRef CAS PubMed.
- R. L. Maher, A. Prasad, O. Rizvanova, S. S. Wallace and D. S. Pederson, DNA Repair, 2013, 12, 964–971 CrossRef CAS PubMed.
- K. Luger, A. W. Mäder, R. K. Richmond, D. F. Sargent and T. J. Richmond, Nature, 1997, 389, 251–260 CrossRef CAS PubMed.
- H. Menoni, M. S. Shukla, V. S. Dimitrov and D. Angelov, Nucleic Acids Res., 2012, 40, 692–700 CrossRef CAS PubMed.
- H. Nilsen, T. Lindahl and A. Verreault, EMBO J., 2002, 21, 5943–5952 CrossRef CAS PubMed.
- J. M. Hinz, Y. Rodriguez and M. J. Smerdon, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4646–4651 CrossRef CAS PubMed.
- A. Prasad, S. S. Wallace and D. S. Pederson, Mol. Cell. Biol., 2007, 27, 8442–8453 CrossRef CAS PubMed.
- V. Singh, B. Kumari, B. Maity, D. Seth and P. Das, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2014, 766, 56–65 CrossRef PubMed.
- B. Wu, K. Mohideen, D. Vasudevan and C. A. Davey, Structure, 2010, 18, 528–536 CrossRef CAS PubMed.
- F. Di Felice, F. Chiani and G. Camilloni, Biochem. J., 2008, 409, 651–656 CrossRef CAS PubMed.
- J. Widom, Q. Rev. Biophys., 2001, 34, 269–324 CrossRef CAS.
- E. Segal, Y. Fondufe- Mittendorf, L. Chen, A. Thastrom, Y. Field, I. K. Moore, J. P. Wang and J. Widom, Nature, 2006, 442, 772–778 CrossRef CAS PubMed.
- H. E. Peckham, R. E. Thurman, Y. Fu, J. A. Stamatoyannopoulos, W. S. Noble, K. Struhl and Z. Weng, Genome Res., 2007, 17, 1170–1177 CrossRef CAS PubMed.
- M. J. Packer, M. P. Dauncey and C. A. Hunter, J. Mol. Biol., 2000, 295, 71–83 CrossRef CAS PubMed.
- V. Iyer and K. Struhl, EMBO J., 1995, 14, 2570–2579 CAS.
- S. Adhikari, A. Uren and R. Roy, J. Biol. Chem., 2008, 2831, 334–339 Search PubMed.
- J. M. Hinz, Y. Rodriguez and M. J. Smerdon, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4646–4651 CrossRef CAS PubMed.
- A. Bacolla, R. Gellibolian, M. Shimizu, S. Amirhaeri, S. Kang, K. Ohshima, J. E. Larson, S. C. Harvey, B. D. Stollar and R. D. Wells, J. Biol. Chem., 1997, 272, 16783–16792 CrossRef CAS PubMed.
- M. A. Young, G. Ravishanker, D. L. Beveridge and H. M. Berman, Biophys. J., 1995, 68, 2454–2468 CrossRef CAS.
- M. M. Greenberg, J. T. Sczepanski and C. Zhou, J. Am. Chem. Soc., 2012, 134, 16734–16741 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra17101b |
| This journal is © The Royal Society of Chemistry 2015 |