
RAFT synthesis of polyethylene glycol (PEG) and amino-functionalized amphiphilic copolymers for dispersing carbon nanofibers†
Zhiwei Yuab,
Kai Xu*a,
Zien Fuab,
Xin Liuab,
Youxiong Zhangab,
Jun Pengab and
Mingcai Chena
aKey Laboratory of Polymer Material for Electronics, Guangzhou Institute of Chemistry, Chinese Academy of Sciences, P.O. BOX 1122, Guangzhou 510650, China. E-mail: xk@gic.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 25th February 2015
Abstract
We designed and synthesized a series of amphiphilic copolymers from poly(ethylene glycol) monoacrylate (PEGMA), 2-(dimethylamino) ethyl methacrylate (DMAEMA), styrene (St), and butyl acrylate (BA) via reversible addition–fragmentation chain transfer (RAFT) polymerization. The dispersion capability of carbon nanofibers (CNFs) by these copolymers as well as their stability were investigated using UV-visible spectroscopy, Fourier transform infrared spectroscopy (FTIR), thermogravimetry (TG), transmission electron microscopy (TEM) and dynamic light-scattering (DLS) techniques. The results show that both of the block copolymers PPEGMA-b-P(DMAEMA-co-St-co-BA) and PDMAEMA-b-P(PEGMA-co-PSt-co-PBA) enabled better efficiencies toward dispersion and functionalization of CNFs compared to the random copolymer and P(PEGMA-co-BA)-b-P(DMAEMA-co-St) polymer, which allowed us to evaluate the effect of block sequences on the dispersing properties and understand the structure–property relationship between CNFs and amino-containing block-copolymers.
1. Introduction
Carbon nanofibers (CNFs) with one-dimensional hollow structure are recognized as unique nanosized fibrous forms of graphitic materials that can be prepared by catalytic decomposition of hydrocarbons over small metal particles. These nanocarbons have interested scientists and engineers because of their widespread potential applications in diverse areas of nanoscience. CNFs have high mechanical strength, well particle erosion resistance and good electrical conductivity, making them ideal reinforcing fibers in composites.1–5 However, most CNFs can easily form aggregates when settled in solutions owing to strong van der Waals interactions between nanofibers. The surface functionalization of CNFs is an essential step for manipulating these nanomaterials. Pristine nanofibers need to be functionalized in order to improve their solubility and be easy for processing. While oxidation of atomic-scale defects or heterochemical doping during growth probably provide the as-synthesized CNFs with relevant functionality, post-synthetic procedures involving incorporation of polar groups are the most convenient and popular strategy to impart hydrophilicity. These modifications can be simply divided into two categories, covalent6,7 and non-covalent functionalizations.8,9 The covalent functionalization of CNFs generally results in a change of carbon hybridization from sp2 to sp3, leading to a possible partial loss of conjugation with the consequence of damaging the desirable mechanical and electrical properties. The advantage of non-covalent functionalization is that chemical functional groups can be introduced onto the surface of CNFs without affecting their graphitic structures.10–12 Non-covalent modification of pristine CNFs in solutions requires additional modifiers or surfactants, including polymeric ones. Physisorption of polymers containing pendent aromatic groups on the CNFs sidewalls, via π–π stacking interaction, allows for a remarkable improvement of dispersion stability in organic and aqueous media and facilitating the preparation of homogeneous CNF-based polymer composites.13,14 Aromatic polymers such as pyrene-based surfactants, poly(phenylene ethynylenes), and heterocyclic conjugated polymer have repeatedly been shown to disperse nanocarbons with high effect of suppressing agglomeration in the literature.15–17 However, the study of amphiphilic copolymers to modify CNFs is relatively insufficient.14,18 Amphiphilic block copolymers that contain amino and phenyl functional groups are frequently used for dispersing inorganic nanoparticles. The lone electron pairs of nitrogen in amino moieties may form a delocalized conjugated system with the sp2-hybridized carbon frameworks. Their basic character offers a possibility whereby one block has a strong affinity for the carbon surface.19 Very recently, several reports demonstrated that PEG block in poly(ethylene glycol)-based amphiphilic surfactant played a crucial role for enhancing the solubilizing and dispersing activity toward carbon nanotubes, and the analogue in composition to CNFs. The highly hydrophilic and flexible PEG chains would extend into solvents, offering repulsion interactions and leading to a stable dispersion of annotates.15,20 In this regard, we have been motivated to evaluate the performance of acrylic copolymer bearing amino and PEG functionalities for dispersing CNFs via non-covalent interactions. The molecular weight, the chain structure and the polymerization sequence were all the key parameters that we wanted to investigate with respect to the functionalization of CNFs. Various controlled radical polymerization methods such as nitroxide-mediated polymerization,21 atom transfer radical polymerization,22 and reversible addition fragmentation chain transfer polymerization (RAFT),23 have been applied for preparation of well-defined polymers with complex architectures including block, graft, and star structures. Among these methods, the RAFT process is probably the most versatile one because of its high degree of compatibility with a wide range of functional monomers.In this paper, we report the synthesis of a series of water-soluble quaternary copolymers from styrene (St), butyl acrylate (BA), N,N-(dimethylamino) ethyl methacrylate (DMAEMA) and methoxy polyethylene glycol monoacrylate (PEGMA) by RAFT polymerization. These copolymers were designed by altering the block sequences. The copolymer-assisted dispersion experiment was performed by adding a defined amount of CNFs to the copolymer aqueous solution, and the results were related to the stability of amphiphilic molecules bound to CNFs. It should be noted that the TEM observation of the carbon residue would give indirect evidence for the copolymer attaching onto nanofibers after drying the CNFs/copolymer suspensions dipped onto copper grids. The adsorption sites of each copolymer and the amounts of aggregated CNFs bundles in aqueous solution may be more difficult to quantify by this method. Finally, carbon black (CB) was used to further study the copolymer performance by sedimentation experiment.
2. Experimental
2.1. Materials and method
N,N-(Dimethylamino)ethyl methacrylate (DMAEMA), styrene (St), butyl acrylate (BA) and 2,2-azoisobutyronitrile (AIBN) were purchased from Aladdin and methoxy polyethylene glycol monoacrylate (PEGMA, SR251) was from Sartomer. All monomers were purified by passing over a basic aluminum oxide column. Azodiisobutyronitrile (AIBN) was crystallized twice from methanol before use. Carbon nanofibers (CNFs, PR-19-XT-PS PU1402) were supplied by Applied Sciences, Inc. This material consists of tubular fibers with a average diameter of about 200 nm and a specific surface area of more than 18 m2 g−1. Carbon black was supplied by Degussa Co. Cyanoisopropyl dithiobenzoate (CyDB) was synthesized according to the literature procedure24 in a modified way (see Fig. S1 and S2 in ESI†).Size exclusion chromatography (SEC) was performed at 35 °C using a Waters 1515 series system equipped with styragel HR4 and HR3 columns and a Waters 2414 refractive index (RI) detector. The system was calibrated with narrowly dispersed polystyrene standards and the mobile phase used was HPLC grade THF flowing at 0.60 mL min−1.
1H NMR spectra was recorded in deuterated chloroform on a Bruker DMX-400 spectrometer equipped with a Varian probe. FTIR spectra were measured on a Bruker Tensor 27 FTIR spectrometer. UV-visible absorption spectra were recorded between 200 nm and 500 nm using a Shimadzu UV-360 PC instrument. Peak intensities of dispersion at 259 nm were read directly from the measured absorption spectra. The thermogravimetric (TG) analysis was performed with a Netzsch TG209F3 thermogravimetric analyzer over a temperature range between 30 and 800 °C at a heating rate of 10 °C min−1 under nitrogen. Transmission electron microscopy (TEM) measurements were performed using a JEM-100CX II microscope at an accelerating voltage of 80 kV. Dynamic light-scattering (DLS) particle sizes were determined on a Malvern Zetasizer instrument.
2.2. Synthesis of random/block copolymers
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). A random copolymer P(PEGMA-co-DMAEMA-co-St-co-BA) denoted as R was obtained and dried under vacuum.
:1). A random copolymer P(PEGMA-co-DMAEMA-co-St-co-BA) denoted as R was obtained and dried under vacuum.Macromolecular RAFT agents (R1). A mixture of BA (1.28 g, 0.010 mol), PEGMA (3.96 g, ∼0.010 mol), RAFT agent (CyDB, 44.2 mg, 0.20 mmol) AIBN (6.50 mg, 0.040 mmol) and 6.0 mL of 1,4-dioxane placed in a 25 mL flask was degassed by three freeze–pump–thaw cycles. The polymerization was then carried out in a constant temperature oil bath at 80 °C. After a reaction time of 2 h, the reaction was stopped by freezing in liquid nitrogen. The P(PEGMA-co-BA) polymerization copolymer denoted as R1 was purified by precipitation into petroleum ether/diethyl ether (mass ratio = 1
:1) and dried under 25 °C vacuum for 24 h.
Block copolymer (B1). AIBN (3.00 mg, 0.018 mmol) and 5.0 mL of 1,4-dioxane were placed into a 25 mL flask followed by addition of St (0.520 g, 0.0050 mol), DMAEMA (0.78 g, 0.0050 mol) monomer and R1 (1.50 g, ∼0.10 mmol). The mixture was degassed by three freeze–pump–thaw cycles. The RAFT polymerization was achieved by heating the mixture to 90 °C for 2 h. Then the reaction flask was immersed immediately into liquid nitrogen. After purification via precipitation into a solvent mixture solvent of petroleum ether and diethyl ether (volume ratio = 1
:1), the product P(PEGMA-co-BA)-b-P(DMAEMA-co-St) denoted as B1 was obtained and dried under 25 °C vacuum for 24 h.
2.3. Dispersion of CNFs in copolymer solution
All CNFs samples were sonicated in acetone for 10 min at 50 W and rinsed with water to remove organic residues as soon as possible. The dispersion was prepared by mixing 0.030 g of CNFs solid with 300 mL of an aqueous copolymer solution (the copolymer concentration is 0.100 mg mL−1) in a flask and then sonicating the resulting mixture for 1 h. CNF/copolymer dispersions were marked as CNF-R, CNF-B1, CNF-B2 and CNF-B3, respectively. All sonication processes were carried out with a horn sonicator. The output power was fixed at 125 W. The flask is placed in a bath of ice water during sonication to prevent rising of the temperature of the mixture.2.4. The dispersion experiment of carbon black
A mixture of carbon black (16.2 g) with acrylate resin (108.1 g), copolymer dispersant propylene glycol methyl ether acetate (8.60 g, mass ratio = 1:1) and xylene/n-butyl acetate mixture solvent (46 g, mass ratio = 5:4) was mechanically milled for 14 h using a shaker ball mill. The obtained CB dispersion was then diluted 100-fold in dimethylbenzene/n-butyl acetate mixture solvent, and subjected to a sonication for 30 min. Samples were denoted as CB followed by the abbreviation of the copolymer used, that is CB-R, CB-B1, CB-B2 and CB-B3, respectively.
3. Results and discussion
3.1. RAFT polymerization
RAFT polymerization can be used to synthesize a wide range of copolymers with controlled architectures and narrow polydispersities. Here, we synthesized copolymers of BA, St, DMAEMA and PEGMA. Random copolymer R is synthesized through one pot reaction, while B1, B2 and B3 were synthesized through two-step solution polymerization by RAFT method (Scheme 1).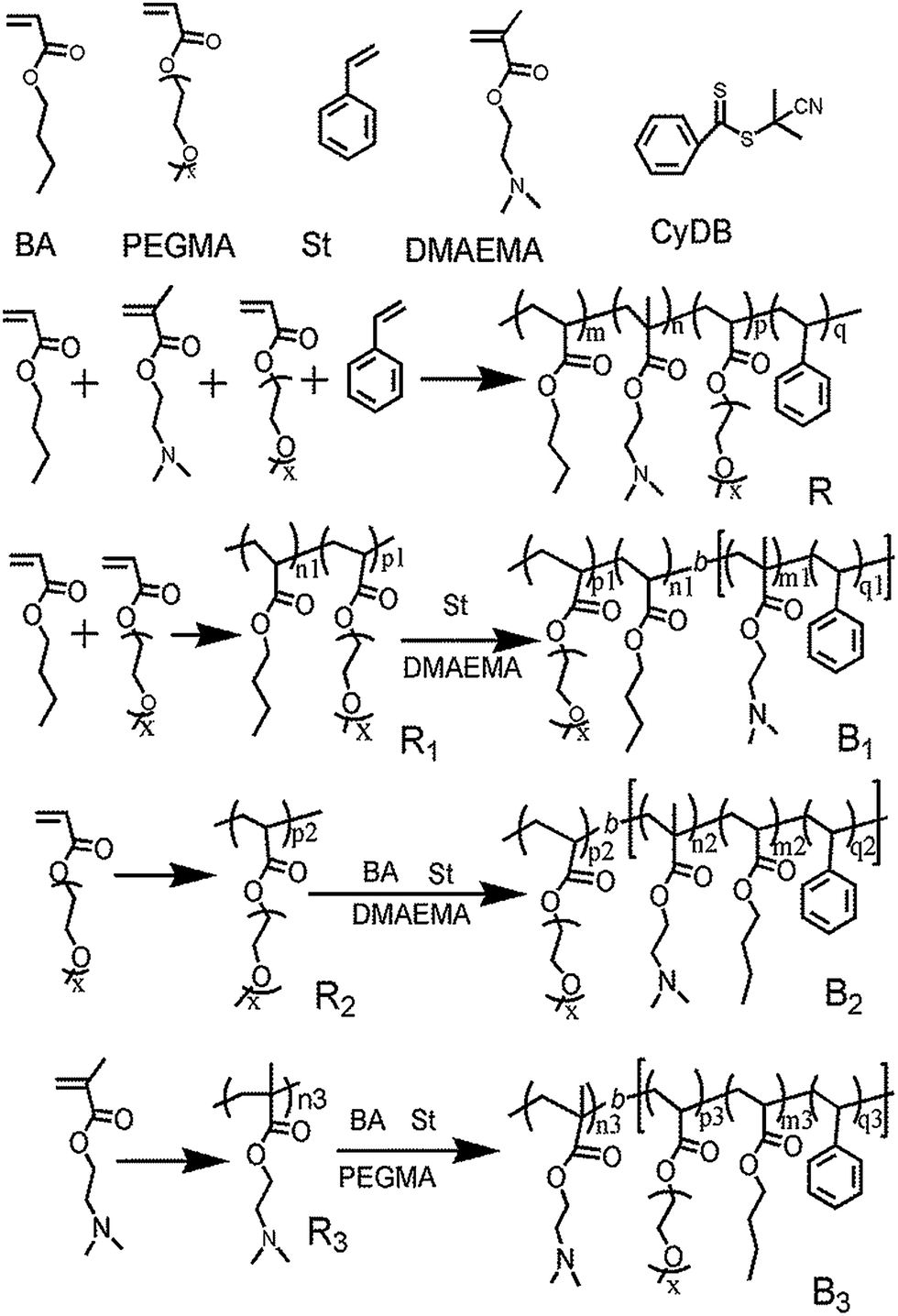 | ||
| Scheme 1 The synthetic routes for copolymers R, B1, B2 and B3, respectively. | ||
The results obtained from SEC were shown in Fig. 1. The values of PDI for all copolymers were in the range of 1.11–1.30, indicating the controllable feature of the polymerization. Elution time of block copolymers was shorter than that of their precursor (Fig. 1b–d), which indicated that block copolymers were successfully synthesized. The PDI of block copolymers had a slight increase compared with their precursor polymers. In addition, there also appeared a very small shoulder on the higher molecular weight side in the SEC traces. We postulated that a combination of the molecular weight distribution of the PEGMA monomer and the occurrence of some chain coupling reactions might account for these results.
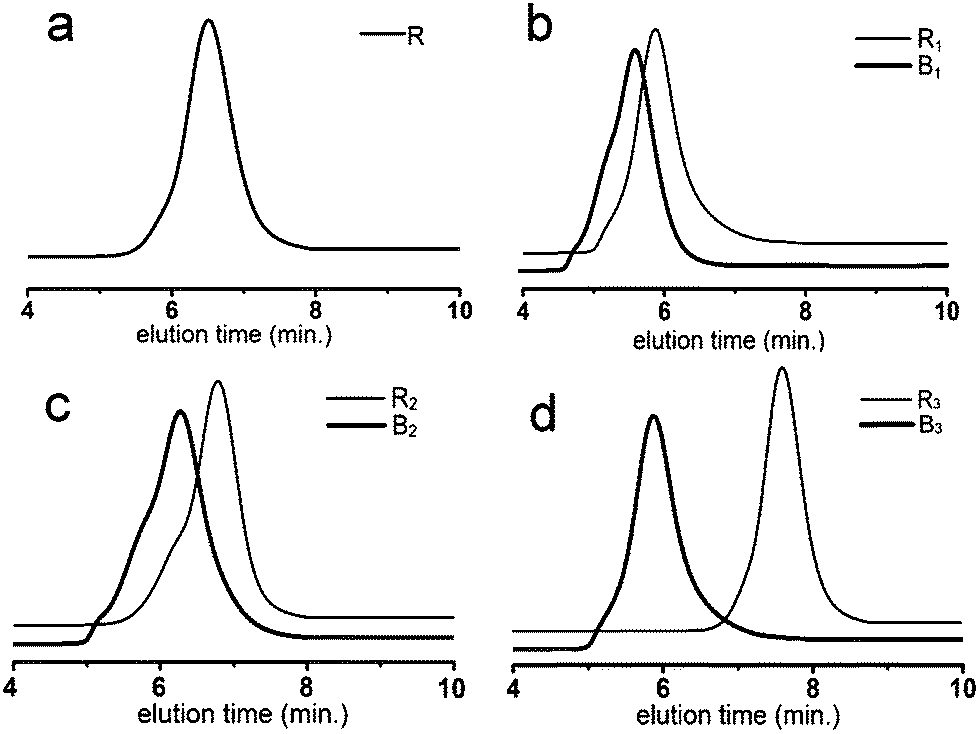 | ||
| Fig. 1 SEC traces of the copolymers and their precursor polymers. | ||
The composition of the copolymer PPEGMA-b-P(DMAEMA-co-St-co-BA) was determined using 1H NMR. Fig. 2 shows a typical 1H NMR spectrum obtained from the precipitated copolymer (B2). The chemical shifts at 3.40 ppm (b) can be assigned to the protons of methoxyl group (–OCH3) of the pendant poly(ethylene glycol) (PEG) chains, demonstrating the existence of PPEGMA block. The characteristic signals at 6.45–7.09 ppm (a) were ascribed to the aromatic protons of the PS block. The protons signals of the methyl (CH3) in the PBA block can be identified at 0.90 ppm (c). The resonance signals at 2–3 ppm (d) corresponded to (CH2N(CH3)2) protons of the PDMAEMA block, which overlapped the characteristic peaks corresponding to the methylene and the methine protons of the backbone. By integrating the signals of (a), (b), (c), and the overlapped signals, we can estimate the mole fraction of each comonomer. The other spectra showed a similar pattern (Fig. S3 in ESI†). The results of the RAFT polymerizations were summarized in Table 1. It must be noted that these results of mole ratio were consistent with the integral calculation of signals at 1–2 ppm from the methylene group of the backbone.
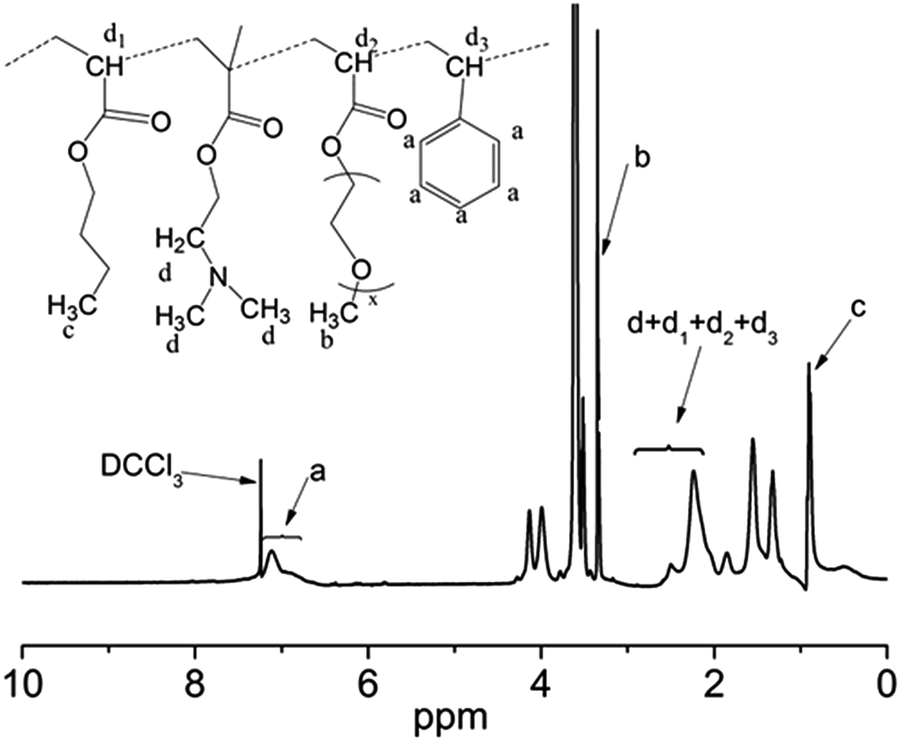 | ||
| Fig. 2 1H NMR spectrum of the copolymer PPEGMA-b-P(DMAEMA-co-St-co-BA). | ||
| Samples | Time (hour) | Temperature (°C) | Total conversiona (±5%) | Mol fractionb (%) | Mnb | Mnc | PDIc Mw/Mn | |
|---|---|---|---|---|---|---|---|---|
| a Total monomer conversion determined gravimetrically.b Mole fraction of four monomers are estimated from 1H NMR spectrum and the ratio order is PEGMA/BA/DMAEMA/St. nd is not determined.c Calculated based on the SEC method. | ||||||||
| R | P(PEGMA-co-DMAEMA-co-St-co-BA) | 4 | 80 | 60 | 27/26/16/31 | 13130 |
11050 |
1.15 |
| R1 | PEGMA-co-PBA | 2 | 80 | 80 | nd | 15250 |
14510 |
1.17 |
| B1 | P(PEGMA-co-BA)-b-P(DMAEMA-co-St) | 2 | 90 | 65 | 28/37/19/17 | 20670 |
19450 |
1.36 |
| R2 | PPEGMA | 2 | 90 | 75 | nd | 10210 |
9190 | 1.15 |
| B2 | PPEGMA-b-P(DMAEMA-co-St-co-BA) | 4 | 90 | 70 | 34/25/23/18 | 17120 |
15210 |
1.30 |
| R3 | PDMAEMA | 6 | 70 | 35 | nd | 3420 | 2390 | 1.15 |
| B3 | PDMAEMA-b-P(PEGMA-co-St-co-BA) | 3 | 90 | 75 | 26/34/9/31 | 17850 |
22340 |
1.21 |
These features, combined with the FTIR evidence (Fig. S4 in ESI†), demonstrated that the quaternary copolymers were successfully synthesized via RAFT method. The four synthesized copolymers were used for dispersing the pristine CNFs in aqueous phase. The pristine CNFs dealt with in this work consisted of loosely aggregated hollow fibers. Their TEM images showed an ordered arrangement of graphene sheets and internal hollow,5 which were responsible for the hydrophobic character of these carbonaceous materials. In the absence of block copolymers, the hydrophobic nanofibers floated on the water surface even or sink in the water with vigorous sonication (Fig. 3, the visual photograph), showing a fairly low dispersibility. Generally, introduction of amine moiety, PEG block and aromatic group in a surface-active composition can enhance the dispersing capacity of carbonaceous materials due to the van der Waals forces, hydrogen bonding and π–π interactions. Indeed, the addition of the synthesized copolymers facilitated the good dispersion of CNFs in water. In other words, the formation of a more stable aqueous dispersion was an evidence that the block copolymers were attached onto the CNFs surface. The attaching ability of our dispersants to assist the nanofiber dispersion was discussed in the next step.
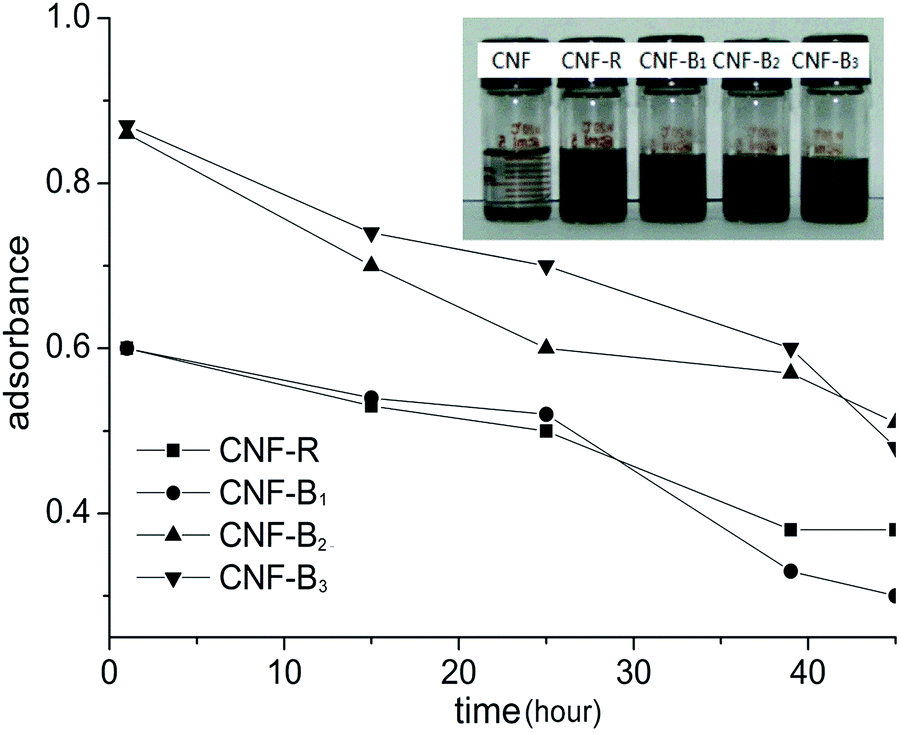 | ||
| Fig. 3 Evolution of the UV-visible absorption curves at 259 nm and the visual observation of the CNFs dispersions. | ||
3.2. UV-visible absorption spectra and stability of CNFs dispersion
The dispersion solutions was usually studied by UV-visible absorption and visual observation.25 As was shown in Fig. 3, it was seen that CNFs could almost not be dispersed in water in the absence of dispersants. From Fig. 3, the absorption of CNF-B2 and CNF-B3 dispersions was more intensive than that of the CNF-R and CNF-B1 dispersion at 259 nm. The result indicated that dispersed in water with the help of in the presence of dispersants B2 or B3 CNFs could disperse more effectively with respect to R or B1.In order to further explore the influence of dispersants on the dispersing properties, UV-visible absorption spectra of the R, B1, B2 and B3 aqueous solutions (Fig. S7 in ESI†) and the supernatants of CNF-R, CNF-B1, CNF-B2 and CNF-B3 dispersions (SCNF-R, SCNF-B1, SCNF-B2 and SCNF-B3, respectively) were measured. The supernatants were obtained at a centrifugal speed of 10000 rpm. SCNF was the supernatant of CNFs dispersion without any dispersant. As shown in Fig. 4, the absorption intensities of the supernatants at 259 nm were all stronger than their corresponding copolymer aqueous solutions. We could calculate and compare the absorption difference values (Da, Db, Dc, and Dd) between the supernatants and their corresponding copolymer aqueous solution at 259 nm, and the increased absorbance intensity might be indicative for the dispersed carbon nanofiber in aqueous media. In addition, the value of Dc and Dd was obviously higher than that of Da and Db. These results therefore seem to suggest that the dispersion of CNF-B2 and CNF-B3 were more stable, with a larger amount of suspended nanofiber under the centrifuging force.
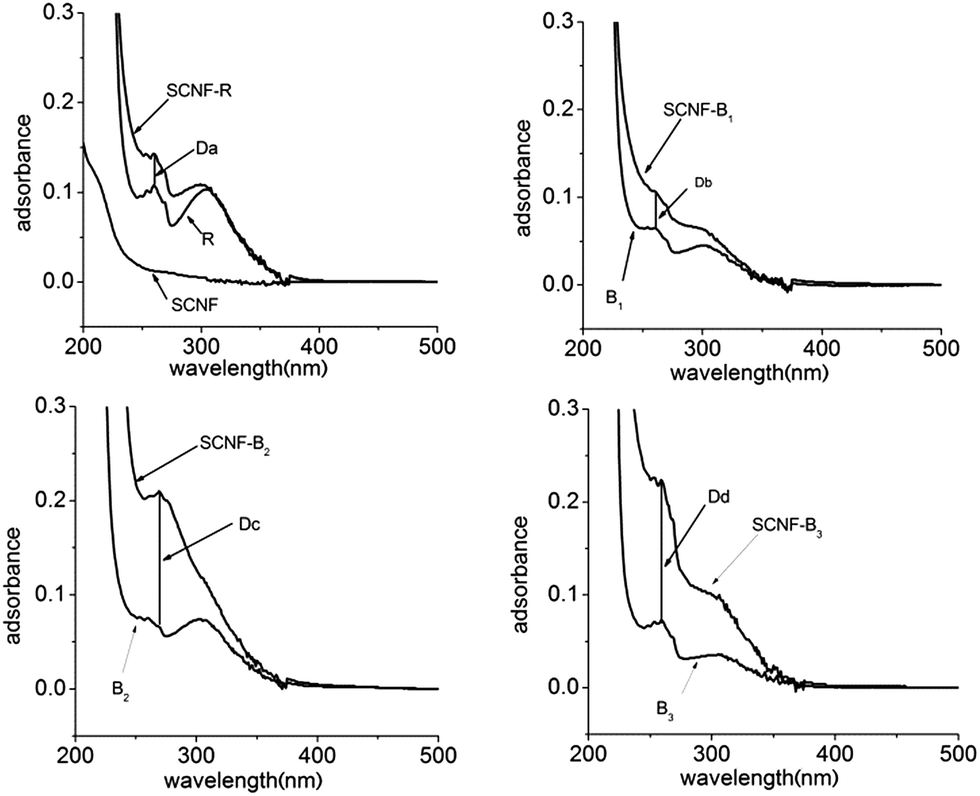 | ||
| Fig. 4 Centrifuged supernatants UV-visible absorption spectra of CNF, CNF-R, CNF-B1, CNF-B2, and CNF-B3 dispersion. | ||
The FTIR technique was highly sensitive and able to examine molecules attached onto the particle surface.12,26,27 CCNF-R, CCNF-B1, CCNF-B2 and CCNF-B3 were the resulting sediment of CNF-R, CNF-B1, CNF-B2 and CNF-B3, respectively, obtained from the centrifugation, and they were sonicated and centrifuged in water for three times. From Fig. 5 and S4,† the copolymers showed strong absorption at 2720–2960 cm−1 and 1720 cm−1; alkyl stretching vibration in the range of 2720–2960 cm−1 and carbonyl vibration absorption peak at 1720 cm−1 can also be observed for the sediments. In addition, characteristic absorption bands in the range of 1000–1400 cm−1 for the sediment samples became sharper compared to that of pristine CNFs, probably originating from the copolymer chains attached onto the CNFs. The adsorption mass of copolymers on the CNFs surface can't be calculated by FTIR, so we took advantage of the thermogravimetric analysis to get the data.
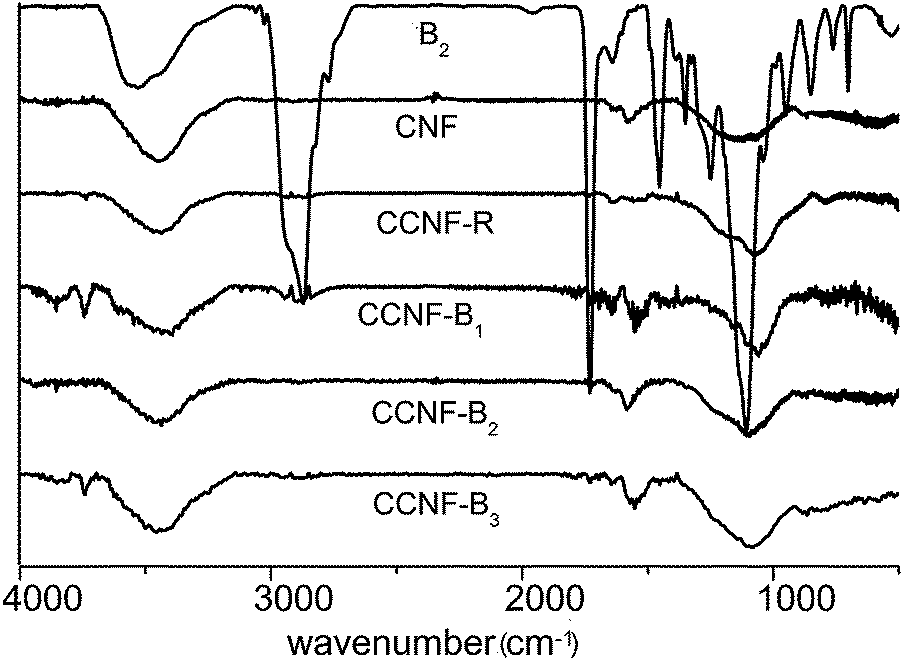 | ||
| Fig. 5 FTIR spectra of sediments of the centrifuged CNF dispersion, pristine CNFs and copolymer. | ||
TG is a very valid tool to figure up dispersant adsorption amount on carbon material surface.28 In Fig. 6, CCNF-R, CCNF-B1, CCNF-B2 and CCNF-B3 obviously had weight loss between 300 and 450 °C while pure CNFs almost had not mass loss in the same temperature range. The inset was the TG curve of B2 (see Fig. S5 for the other copolymers in ESI†), which decomposed completely between 300 and 450 °C. Hence, we can deduce that the weight loss of CCNF-R, CCNF-B1, CCNF-B2 and CCNF-B3 between 300 and 450 °C was caused by the decomposition of the copolymers adsorbed on the CNFs surface, and they were 4.7%, 6.8%, 3.7% and 3.1%, respectively.
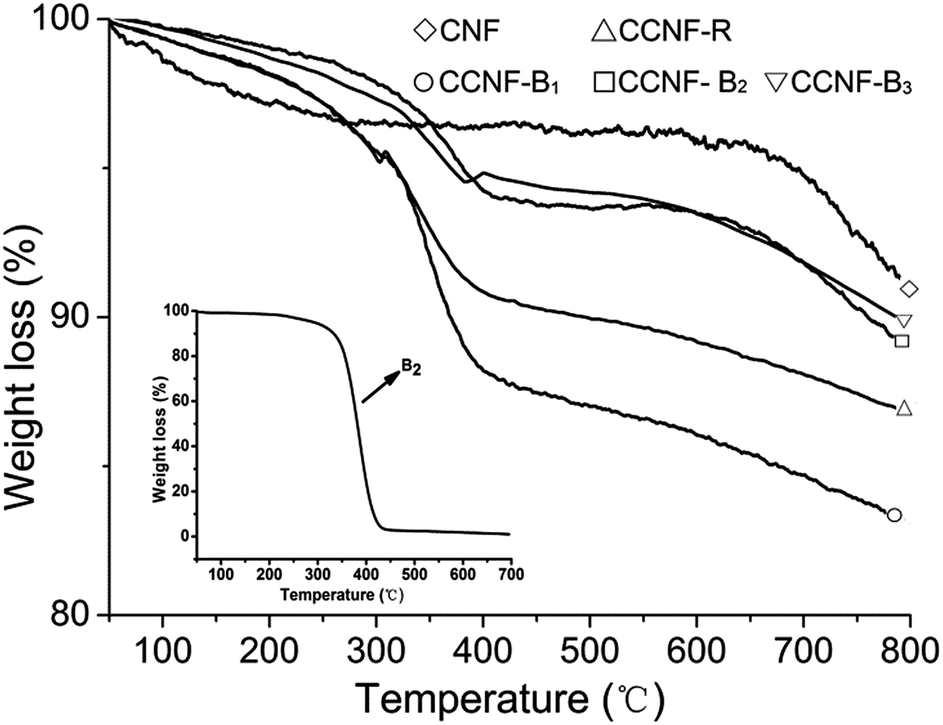 | ||
| Fig. 6 The TG plots of the centrifugal sediments of the CNFs dispersion. | ||
3.3. Characterization of CNFs dispersions by TEM and DLS
Transmission electron microscopy (TEM) images could provide evidence for the dispersion of CNFs suspension particles as shown in Fig. 7, because TEM observations carried out at different magnifications could reveal the existence of CNFs with various diameters and lengths. CNFs usually aggregate and entangle by the way of large ropes without any modification. By adding R, B1, B2 and B3 to modify CNFs, respectively, the CNFs apparently tend to exist as individuals or very small bundles, which was in sharp contrast to the case of raw materials. | ||
| Fig. 7 TEM of CNFs dispersion. (A) and (a) CNF-R, (B) and (b) CNF-B1, (C) and (c) CNF-B2, (D) and (d) CNF-B3. | ||
For CNFs dispersed by copolymers, small bundles were frequently seen in the dispersion. This is because the anchor parts of the copolymers also interact with the sidewalls of adjacent CNFs, not only individual sticks.17 Expectedly, in Fig. 7a–d, from a survey of many areas of the specimen it was evident that copolymers were evenly distributed over the nanofiber surface (labelled by the arrow and circle).
To further analyze the dispersion of CNFs in aqueous solution, we use DLS to estimate the size distribution of CNFs in different copolymer solutions. DLS data did not directly show the CNFs diameters quantitatively for its nature, because if the particle was long, the data would include the effect of the length. As shown in Fig. 8, the main size distribution of CNFs in CNF-B2 or CNF-B3 was between 500 and 1050 nm, and that in CNF-B2 or CNF-B3 was between 600 and 1200 nm. The differences of the distribution indicated, most likely, CNF-R or CNF-B1 had more CNFs bundles in comparison with CNF-B2 or CNF-B3. The DLS result of the pure CNFs was in the ESI (Fig. S6†). From the DLS analysis, it can be concluded that CNFs modified by copolymers exists mainly in the form of individual sticks in aqueous solution as well as in small bundles and, with addition of B2 or B3 the dispersing capability for CNFs improved more effectively than with R or B1.
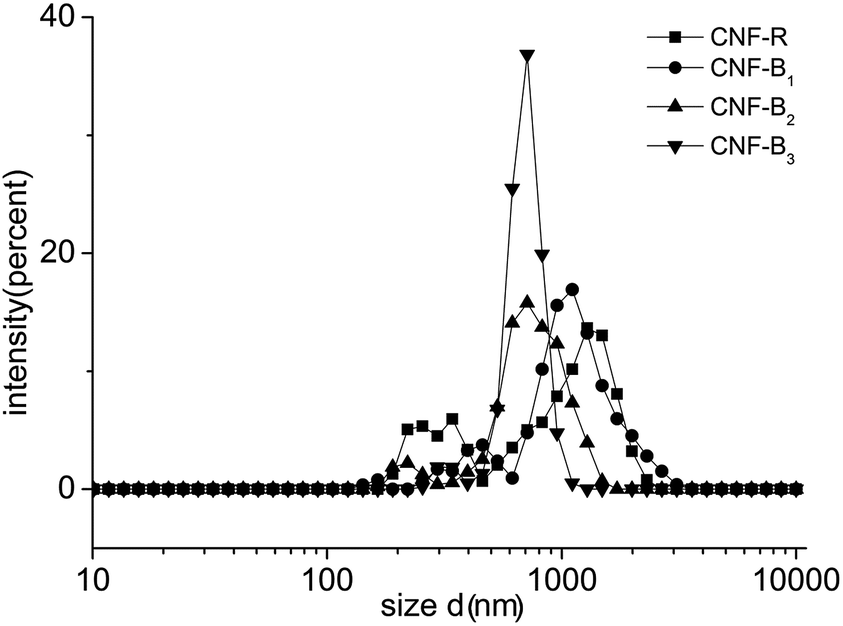 | ||
| Fig. 8 The size distributions of CNFs dispersions by DLS. | ||
Some special copolymers could impede CNFs aggregation mainly by well-known steric repulsion.29,30 When CNFs were mixed with our copolymers, the PDMAEMA, PSt, and PBA segments would interact with the sidewall of the CNFs (the interaction was often called “coat” or “decorate”) while methoxy polyethylene glycol groups from the PPEGMA would extend into water to form “buoys” parts and hence create a so-called steric repulsion.31,32 (Scheme 2) In this paper, for non-covalent functionalization of CNFs, B2 and B3 copolymers, which contained a PPEGMA and PDMAEMA homopolymer segment respectively, showed a little better dispersing performance than R and B1 possessing two random copolymers. For B2, PPEGMA could form stable steric hindrance in aqueous solution and for B3, PDMAEMA segment could form a uniform and stable adsorption layer on the surface of the CNFs. The homopolymer segments were beneficial for CNFs functionalization because of those special roles. However, R or B1 cannot form an adsorbed layer through the same process. Therefore, B2 or B3 present better dispersing properties for CNF in aqueous solution, even though the side chains of all copolymers contained polyethylene glycol (PEG) and amino.
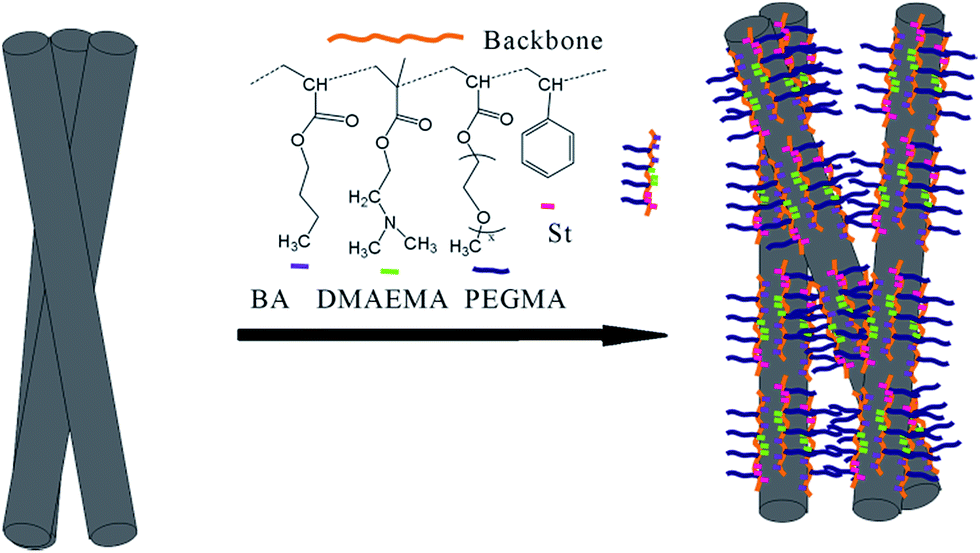 | ||
| Scheme 2 Interaction between the copolymers and CNFs. | ||
To further verify the copolymer dispersing performances, CB was used to be functionalized through sedimentation experiment. In this part, the functionalization of the carbon black was also difficult. CB was mixed with the synthesized copolymers in acrylate resin, and then carbon black dispersions were diluted by solvents to observe their dispersing performance. As shown in Fig. 9, sample CB-R and CB-B1 obviously became clearer 5 days later, while CB-B2 and CB-B3 almost had no change. After one month, CB-R and CB-B1 almost became clear, but CB-B2 and CB-B3 remained as a stable dispersion.
 | ||
| Fig. 9 CB sedimentation pictures I (0 day), II (after 5 days) and III (after one month). | ||
4. Conclusions
Copolymers of St, BA, DMAEMA and PEGMA were successfully synthesized by reversible addition fragmentation chain transfer (RAFT) polymerization. The molecular weight and PDI of the copolymers were determined by SEC and the structure by 1H NMR. The PDIs of copolymers obtained were between 1.14 and 1.36. The dispersing performances of the functionalizing CNFs by the copolymers were measured by UV, FTIR, TG and TEM. The copolymers can well modify CNFs against aggregation mainly due to the steric repulsion. The characterization results also demonstrated that the dispersing performances of copolymer B2 or B3 were better than R or B1. This was probably due to that both of B2 or B3 bore a specific homopolymer segment, which was beneficial to the dispersion of CNFs, even though the side chains of all the copolymers contained the polyethylene glycol (PEG) and amino. Finally we studied their performances by modifying carbon black and got almost the same results that B2 or B3 had better dispersibility for CB. In the next study, we will further research the CNFs or other materials modified by this kind of copolymers. Meanwhile, we are also considering quasi-block copolymers by one pot RAFT synthesis33,34 from the view points of practical application and economy.Acknowledgements
This research was supported by the Guangdong Province Research Project (2011A091000007). And we are also grateful to the National Natural Science Foundation of China for its financial support (Registry no. 21174162).References
- J. P. Kim, Y. R. Noh, J.-U. Kim and J.-S. Park, Thin Solid Films, 2011, 519, 7899–7903 CrossRef CAS PubMed.
- J. Zhu, L. Mu, L. Chen, Y. Shi, H. Wang, X. Feng and X. Lu, Macromol. Chem. Phys., 2014, 14, 1407 CrossRef.
- R. L. Price, M. C. Waid, K. M. Haberstroh and T. J. Webster, Biomaterials, 2003, 24, 1877–1887 CrossRef CAS.
- J. Tatami, T. Katashima, K. Komeya, T. Meguro and T. Wakihara, J. Am. Chem. Soc., 2005, 88, 2889–2893 CAS.
- J. Wu, H. Cai, K. Xu, Z. Fu, X. Liu and M. Chen, J. Mater. Chem. C, 2013, 1, 646–656 RSC.
- O. V. Kharissova, B. I. Kharisov and E. G. de Casas Ortiz, RSC Adv., 2013, 3, 24812–24852 RSC.
- J. Zhu, J. Hiltz, R. B. Lennox and R. Schirrmacher, Chem. Commun., 2013, 49, 10275–10277 RSC.
- K. Saint-Aubin, P. Poulin, H. Saadaoui, M. Maugey and C. Zakri, Langmuir, 2009, 25, 13206–13211 CrossRef CAS PubMed.
- Z. Song, J. Dai, S. Zhao, Y. Zhou, F. Su, J. Cui and Y. Yan, RSC Adv., 2014, 4, 2327–2338 RSC.
- N. Karousis, N. Tagmatarchis and D. Tasis, Chem. Rev., 2010, 110, 5366–5397 CrossRef CAS PubMed.
- S. W. Kim, T. Kim, Y. S. Kim, H. S. Choi, H. J. Lim, S. J. Yang and C. R. Park, Carbon, 2012, 50, 3–33 CrossRef CAS PubMed.
- Y. Nie and T. Hübert, Composites, Part A, 2012, 43, 1357–1364 CrossRef CAS PubMed.
- O. V. Kharissova, B. I. Kharisov and E. G. de Casas Ortiz, RSC Adv., 2013, 3, 24812–24852 RSC.
- J. Zou, S. I. Khondaker, Q. Huo and L. Zhai, Adv. Funct. Mater., 2009, 19, 479–483 CrossRef CAS.
- A. Di Crescenzo, M. Aschi and A. Fontana, Macromolecules, 2012, 45, 8043–8050 CrossRef CAS.
- X. Xin, G. Xu, T. Zhao, Y. Zhu, X. Shi, H. Gong and Z. Zhang, J. Mater. Chem. C, 2008, 112, 16377–16384 CAS.
- G. Yu, M. Xue, Z. Zhang, J. Li, C. Han and F. Huang, J. Am. Chem. Soc., 2012, 134, 13248–13251 CrossRef CAS PubMed.
- H. Sawada, N. Naitoh, R. Kasai and M. Suzuki, J. Mater. Sci., 2008, 43, 1080–1086 CrossRef CAS PubMed.
- M. W. West, U.S. Patent, 4,755,563, 1988.
- M. Granite, A. Radulescu, W. Pyckhout-Hintzen and Y. Cohen, Langmuir, 2010, 27, 751–759 CrossRef PubMed.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- S. H. Thang, R. T. Mayadunne, G. Moad and E. Rizzardo, Tetrahedron Lett., 1999, 40, 2435–2438 CrossRef CAS.
- S. Attal, R. Thiruvengadathan and O. Regev, Anal. Chem., 2006, 78, 8098–8104 CrossRef CAS PubMed.
- L. Jiang, L. Gao and J. Sun, J. Colloid Interface Sci., 2003, 260, 89–94 CrossRef CAS.
- J. Wu, H. Cai, K. Xu, Z. Fu, X. Liu, M. Chen and X. Zhang, Chem. Phys. Lett., 2012, 522, 78–83 CrossRef PubMed.
- N. Grossiord, P. van der Schoot, J. Meuldijk and C. E. Koning, Langmuir, 2007, 23, 3646–3653 CrossRef CAS PubMed.
- A. Star, D. W. Steuerman, J. R. Heath and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 2508–2512 CrossRef CAS.
- X. Xin, G. Xu, T. Zhao, Y. Zhu, X. Shi, H. Gong and Z. Zhang, J. Phys. Chem. C, 2008, 112, 16377–16384 CAS.
- P. D. Petrov, G. L. Georgiev and A. H. Müller, Polymer, 2012, 53, 5502–5506 CrossRef CAS PubMed.
- Y. Sun, W. Fu, Z. Li and Z. Wang, Langmuir, 2014, 30, 8615–8620 CrossRef CAS PubMed.
- C. Guerrero-Sanchez, L. O'Brien, C. Brackley, D. J. Keddie, S. Saubern and J. Chiefari, Polym. Chem., 2013, 6, 1857–1862 RSC.
- J. J. Haven, C. Guerrero Sanchez, D. J. Keddie and G. Moad, Macromol. Rapid Commun., 2014, 35, 492–497 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra15925j |
| This journal is © The Royal Society of Chemistry 2015 |