
Intramolecular cyclization assisted oxidative addition: synthesis of octahedral cycloplatinated methyl complexes†
D. Shankara,
K. Jaipalb,
B. Sridharc,
R. Nagarjuna Charyd,
S. Prabhakard,
L. Giribabu*b and
Pravin R. Likhar*a
aOrganometallic Gp., Inorganic and Physical Chemistry Division, CSIR-Indian Institute of Chemical Technology, Hyderabad-500007, India. E-mail: plikhar@iict.res.in; Fax: +91-040-27160921; Tel: +91-040-27191667
bSolar Cell Gp., Inorganic and Physical Chemistry Division, CSIR-Indian Institute of Chemical Technology, Hyderabad-500007, India. E-mail: giribabu@iict.res.in; Fax: +91-040-27160921; Tel: +91-040-27191724
cX-ray Crystallography Centre, CSIR-Indian Institute of Chemical Technology, Hyderabad-500007, India
dNational Centre for Mass Spectrometry, CSIR-Indian Institute of Chemical Technology, Hyderabad-500007, India
First published on 9th February 2015
Abstract
Cycloplatination of XMeC6H4C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CC-(Rf) (
CC-(Rf) (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N-4-OCH3C6H4) [X = S, Se and Rf = perfluoroalkyl groups] with PtCl2 gave a monomeric platinum(IV) complex having an octahedral structure with one annulated heterocycle coordinated in a plane and another out of the plane. The formation of platinum(IV) complex 2a, confirmed by analysis of its X-ray crystal structure, follows simultaneous cyclization and oxidative addition of methyl chloride generated during the cyclization. The emission and reactivity studies of platinum(IV) complexes are discussed.
N-4-OCH3C6H4) [X = S, Se and Rf = perfluoroalkyl groups] with PtCl2 gave a monomeric platinum(IV) complex having an octahedral structure with one annulated heterocycle coordinated in a plane and another out of the plane. The formation of platinum(IV) complex 2a, confirmed by analysis of its X-ray crystal structure, follows simultaneous cyclization and oxidative addition of methyl chloride generated during the cyclization. The emission and reactivity studies of platinum(IV) complexes are discussed.
Introduction
The oxidative addition reaction is one of the most fundamental processes in catalytic reactions initiated by transition metals.1 Although the oxidative addition of Pd0 to PdII is a well established process2 compared to PdII/PdIV, numerous reports on the potential involvement of PdIV intermediates have been proposed in catalysis and organic synthesis without definite evidence.3,4 In contrast to palladium chemistry, two electron oxidation and reduction reactions between Pt0, PtII and PtIV are an integral part of platinum chemistry and platinum, in lower oxidation state, dominates the oxidative reaction to its higher oxidation states.5 In this context, σ-alkyl complexes of platinum(II) have been thoroughly studied and it has been demonstrated that coordinatively unsaturated 16-electron organoplatinum(II) complexes are highly amenable for oxidative addition process to afford stable and inert coordinatively saturated 18-electron organoplatinum(IV) products.6 Furthermore, the investigations suggest that diimine based nitrogen-donor ligands are far better than phosphine and arsine ligands in stabilizing the organoplatinum(IV) products due to the favourable electronic and steric properties of the nitrogen-donors.7It has also been well studied that platinum(II) diimine complexes (2,2′-bypyridine or 1,10-phenanthroline) are extremely reactive towards the oxidative addition of alkyl halides8 and the oxidative addition of aryl halides can only takes place if the diimine ligand contains a pendant aryl–halogen bond (intramolecular oxidative addition).9 In contrast to these reports, the examples on the intramolecular oxidative addition of alkyl/aryl halides are very rare.10
In our previous findings, we studied the intramolecular cyclizations of o-XMeC6H4CCC-(CF3)(N-4-OCH3C6H4) where X = S and Se, with PdCl2(PhCN)2 to dimeric cyclopalladated benzothiophene and benzoselenophene.11 Further, by changing the imine moiety with pyridine, we could successfully isolate the SCN pincer-type palladacycles.12 The promising biological activities of palladacycles and their reactivity towards electrophiles encouraged us to investigate the further reactivity of propargyl imines with platinum metal. In continuation to our research interest in the synthesis of different metallocycles, we report here a cyclizations and platinacyclization of o-XMeC6H4CCC-(Rf)(N-4-OCH3C6H4) where X = S, Se and Rf = perfluoroalkyl groups with PtCl2 to yield highly stable octahedral bis-ligated platinacycles. Interestingly, in the formation of octahedral platinacycles, Pt(II) get oxidise to Pt(IV) by oxidative addition of methyl group which was generated during the intramolecular cyclization.
Results and discussion
Ortho thioanisole and ortho selenoanisole substituted perfluoroalkyl propargyl imines were prepared according to previously published results.13 The alkynes 1a, 1b, 1c, and 1d were obtained by palladium promoted Sonogashira coupling of corresponding perfluoroalkyl imidoyl iodides and the corresponding alkynes.14 All the imines were fully characterised by 1H NMR, 13C NMR and mass spectroscopy. Various platinum(II) salts were used to react with ortho thioanisole substituted perfluoromethyl propargyl imine 1a to synthesise platinacycle (2a). However, the best yield of 2a was achieved with platinum(II) dichloride (Scheme 1). Thus, the reaction of ortho thioanisole substituted perfluoromethyl propargyl imine with excess PtCl2 in dry toluene at room temperature for 24 h under N2 atmosphere afforded air and water stable crude platinacycle as dark red solid which on subjected to column chromatography yielded 70% pure platinacycle (2a).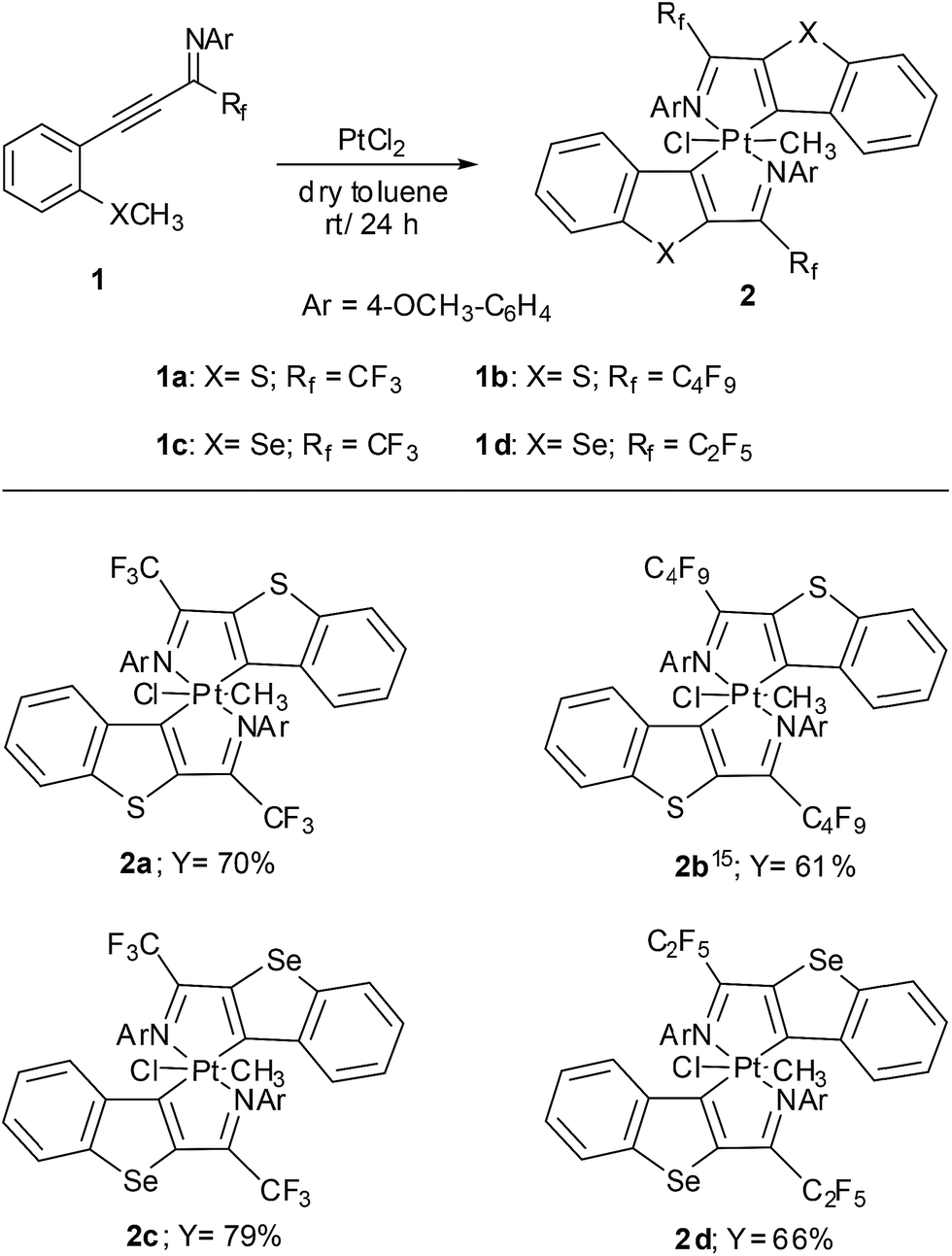 | ||
| Scheme 1 Preparation of platinacycle 2. | ||
Though the preparation of all the platinacycles (2a, 2b, 2c and 2d) were attempted at room temperature under rigorous nitrogen atmosphere, the aerobic atmosphere of the reaction afforded desired platinacycles along with inseparable unidentified compounds. In case of 2b,15 the reaction of bulky perflurobutyl propargyl imine (1b) with Pt(II)Cl2 proceeded at high temperature (110 °C) with longer reaction time and afforded lesser yield compared to other platinacycles. The requirement of high temperature and longer reaction time might be due to the presence of bulky perflurobutyl group.
All the platinacycles were characterised by 1H NMR, 13C NMR, IR and CHN analysis. The 1H NMR spectra of platinacycles 2 containing S and Se shows two singlet peaks in the range of 3.6–3.9 and 13C NMR spectra shows peaks at 55.2 and 55.7 ppm correspond to methoxy group attached to phenyl ring. The 1H NMR for methyl protons directly bonded to platinum atom shows a singlet with satellite peaks at around 1.97 to 2.20 ppm and 13C NMR shows around −6 to −7 ppm. Whereas all the propargyl imine ligands 1 show singlet peak at the range of 3.8–3.9 and 2.5 ppm corresponding to methoxy group attached to phenyl ring and methyl protons attached to sulphur or selenium. The 1H NMR spectra of 2 clearly shows the absence of CH3 signal of o-SCH3C6H5 group after cyclization. Further, the change in chemical shift of 13C NMR of imine carbon atom (CN) from 134 ppm to 159 ppm suggests the interaction of nitrogen to the platinum atom.16 The protons from the methyl group bonded to platinum center appeared at slightly down field (approx. 2.00 ppm) when compared to the proton from other methyl–platinum complexes (1.4–1.7 ppm). In infrared study, the characteristic skeletal vibrations for alkyne ligands 1 were observed in the range of 2190–2000 cm−1 which were absent after the formation of platinacycle 2.
The dark reddish-yellow single crystal suitable for the X-ray single crystal study was obtained by slow evaporation of platinacycle 2a solution (CH3OH/EtOAc in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) in air after three weeks at room temperature. Platinacycle 2a crystallizes in a monoclinic space group P21/n and clearly reveals the formation of a bis-ligated platinum(IV) with chloride ion and methyl group in an octahedral fashion. The C24 and N2 of the imine substituted benzothiophene is approximately planar with Pt1, whereas C8 and N1 of the other imine substituted benzothiophene adopts axial (planar with Pt1) and equatorial position with significant decrease in bond angle of C8–Pt1–N1 to 79.63(16). The angles between adjacent atoms in the coordination sphere of platinum lie in the range 79.63–177.32 (Fig. 1).
:50) in air after three weeks at room temperature. Platinacycle 2a crystallizes in a monoclinic space group P21/n and clearly reveals the formation of a bis-ligated platinum(IV) with chloride ion and methyl group in an octahedral fashion. The C24 and N2 of the imine substituted benzothiophene is approximately planar with Pt1, whereas C8 and N1 of the other imine substituted benzothiophene adopts axial (planar with Pt1) and equatorial position with significant decrease in bond angle of C8–Pt1–N1 to 79.63(16). The angles between adjacent atoms in the coordination sphere of platinum lie in the range 79.63–177.32 (Fig. 1).
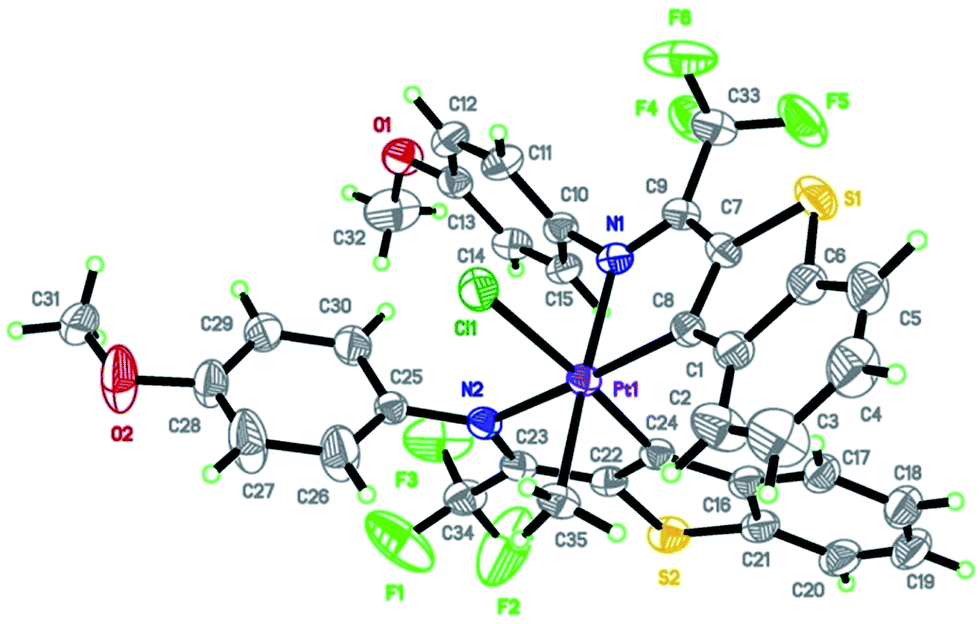 | ||
| Fig. 1 The molecular structure of 2a, with the atom-numbering scheme. Displacement ellipsoids are drawn at the 30% probability level and H atoms are shown as small spheres of arbitrary radius. Selected bond lengths [Å] and bond angles [°]: N1–Pt1 2.194(3), N2–Pt1 2.152(3), C8–Pt1 2.017(4), C24–Pt1 2.008(4), C35–Pt1 2.090(4), Cl1–Pt1 2.3884(11); C8–Pt1–N1 79.63(16), C24–Pt1–N2 79.82(15), C35–Pt1–N1 177.32(15), C35–Pt1–Cl1 93.39(14). | ||
The concerted intramolecular oxidative addition has been rarely known for the organoplatinum(II) complexes in the literature.17,10 The dissociation of one of the amine group prior to oxidative addition through ortho-metalation is the key step in the synthesis of Pt(IV) complexes. In the formation of platinacycle 2a, we speculate that the first unit of 1a initially reacts with Pt(II)Cl2 through coordination of imine and alkyne bond with elimination of one equivalent methyl chloride.11b In the later stage, second unit of 1a reacts with intermediate B in a similar fashion to form intermediate C. The coordinatively unsaturated Pt(II) intermediate C tends to undergo oxidative addition by methyl chloride present in the reaction mixture to form a stable octahedral platinacycle coordinated by two diimines nitrogen, two benzothiophenes (at C3-position) and each chloride ion and methyl group (Scheme 2).
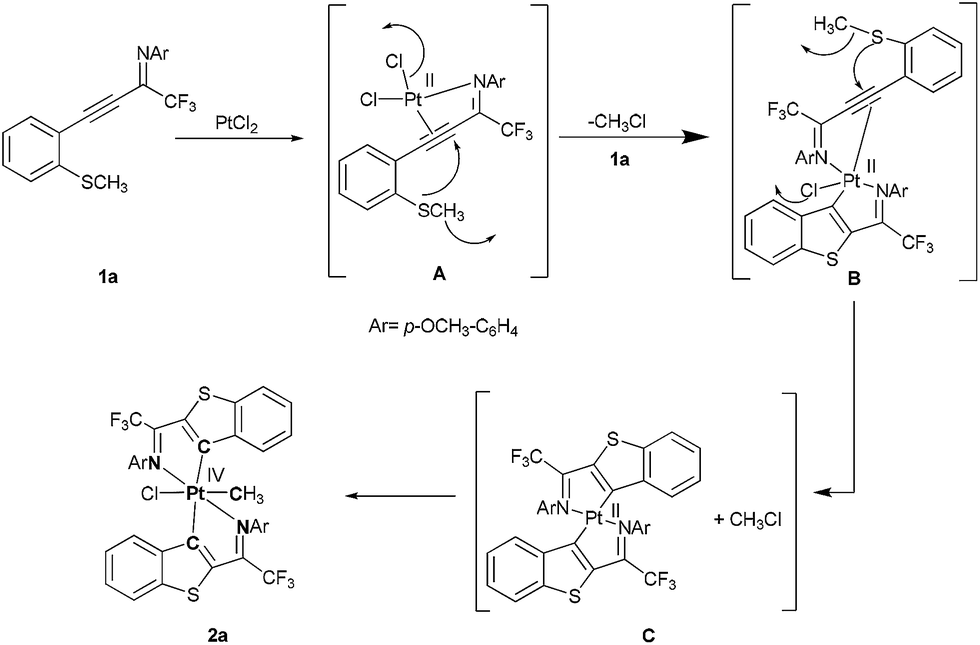 | ||
| Scheme 2 Plausible reaction mechanism for the formation of platinacycle. | ||
In situ ESI mass spectrometry experiments were performed on the reaction mixture of propargyl imine 1a and platinum dichloride (reaction was terminated at 12 h) to investigate the species generated during the reaction. It is easy to identify the Pt incorporated species in the mass spectrum by the specific isotopic pattern of Pt. The positive ion ESI mass spectrum (Fig. 1, ESI†) showed platinum incorporated species above m/z 550, which indicates that only propargyl imine reacted (decomposition/dimerization) under given experimental conditions. The spectrum included three Pt containing ions; one major ion at m/z 878.09 (mass value of highest isotope) and two other abundant ions at m/z 602.07 and 638.04. The ion at m/z 878.09 identified as the species corresponding to platinum with bis-ligand and one methyl group (similar to the species 2a in Scheme 2 without chlorine). This is confirmed based on accurate mass values and isotopic pattern distribution (Table 1, ESI†). The identification of m/z 878.0894 species suggests that Pt(III) must be generated through the square planer Pt(II), C intermediate. The ion at m/z 602.07 matched to a platinum complex with one ligand, CH3CN and CH3OH (see ESI†). We assume that this ion is formed by replacement of two labile chlorides of PtCl2 with cyclized propargyl imine, CH3OH and CH3CN (solvents used in ESI† analysis). We could not propose a structure for the ion appeared at m/z 638.04.
| Compounds | Absorption (nm) | Emission (nm) | Red shift (nm) |
|---|---|---|---|
| 1a | 270, 351 | 310, 353, 372 | 40 |
| 1b | 227, 269 | 310, 354, 374 | 39 |
| 1c | 269, 355 | 310, 353, 372 | 39 |
| 1d | 228, 270, 345 | 310, 353, 373 | 40 |
| 2a | 258, 371 | 425 | 54 |
| 2b | 259, 380 | 435 | 55 |
| 2c | 270, 373 | 438 | 65 |
| 2d | 260, 380 | 436 | 56 |
The formation of platinacycle 2a was investigated by absorption study. Initially, the absorption spectra for the reaction of ligand 1a with Pt(II) Cl2 was studied at time t = 0; the UV-visible spectral changes occurred during the formation of 2a are shown in Fig. 2. From Fig. 2, it was noticed that there was an increase in absorption peak near 260 nm and a decrease in absorption peak near 230 nm in the complex 2a when compared to ligand 1a which indicates increase in conjugation of ligand (hetero-annulation of ligand 1a) in the UV region. This absorption band indicates that bis-ligated platinum complex with two discrete ligands bound to one metal centre favours high conjugation to yield the peak position at lower energy and this band can traditionally be assigned as MLCT (Metal–Ligand Charge Transfer).18
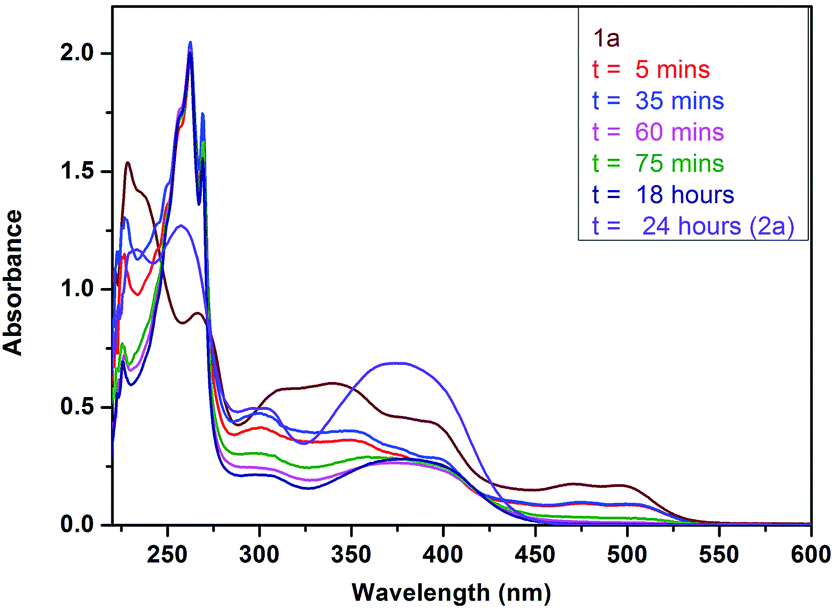 | ||
| Fig. 2 In situ UV-vis spectral changes during the formation of Pt(IV) complex 2a at reaction time = 5 min, 35 min, 60 min, 75 min, 18 h, 24 h. in CH2Cl2 solution (1.6 × 10−5 M). | ||
We have observed an isosbestic point at 350 nm. All the ligands 1a, 1b, 1c and 1d do not absorb the light in the visible region (see ESI†), while the complexes 2a, 2b, 2c and 2d exhibit well-defined low energy absorption peak at 360–400 nm (near visible region) (Fig. 3).
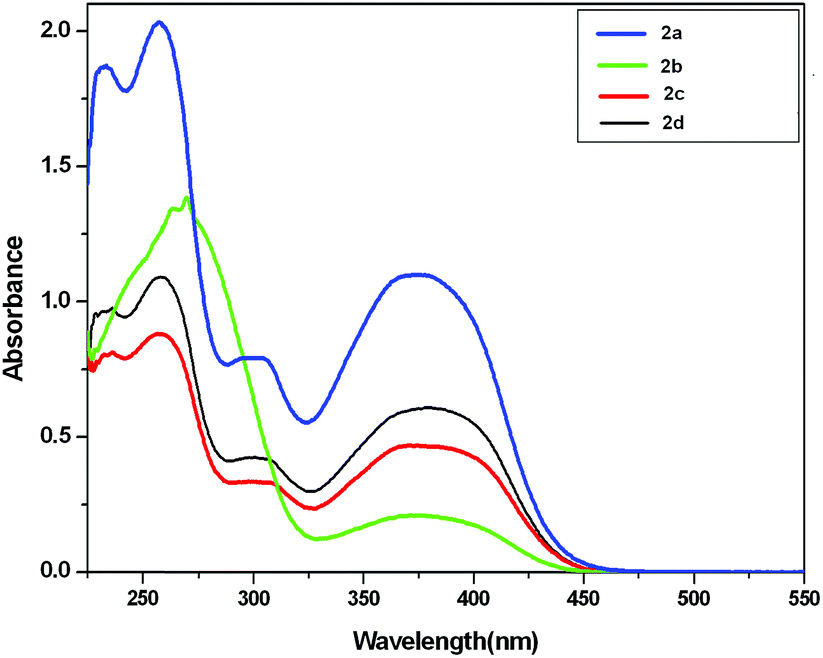 | ||
| Fig. 3 UV-vis spectra of Pt(IV) complexes 2a, 2b, 2c and 2d in CH2Cl2 solution (2.4 × 10−4 M). | ||
The redox properties of ligands and their platinum complexes were studied by cyclic and differential pulse voltammetry in CH3CN containing 0.1 M TBAPF6 at room temperature. Fig. 4 depicts representative differential pulse voltammogram of ligand 1a and complex 2a. The ligand 1a showed one reversible oxidation wave at 1.72 V and two either reversible or quasireversible reduction potentials at −1.04 V and −1.47 V, while complex 2a has also shown oxidation wave at 1.75 V and two reduction potentials at −1.03 V and −1.47 V. From the CV and DPV study, it can be concluded that both the ligands and complexes undergo either reversible or quasireversible oxidation and reduction reactions. We have observed similar electrochemistry in other ligands and complexes (see ESI†).
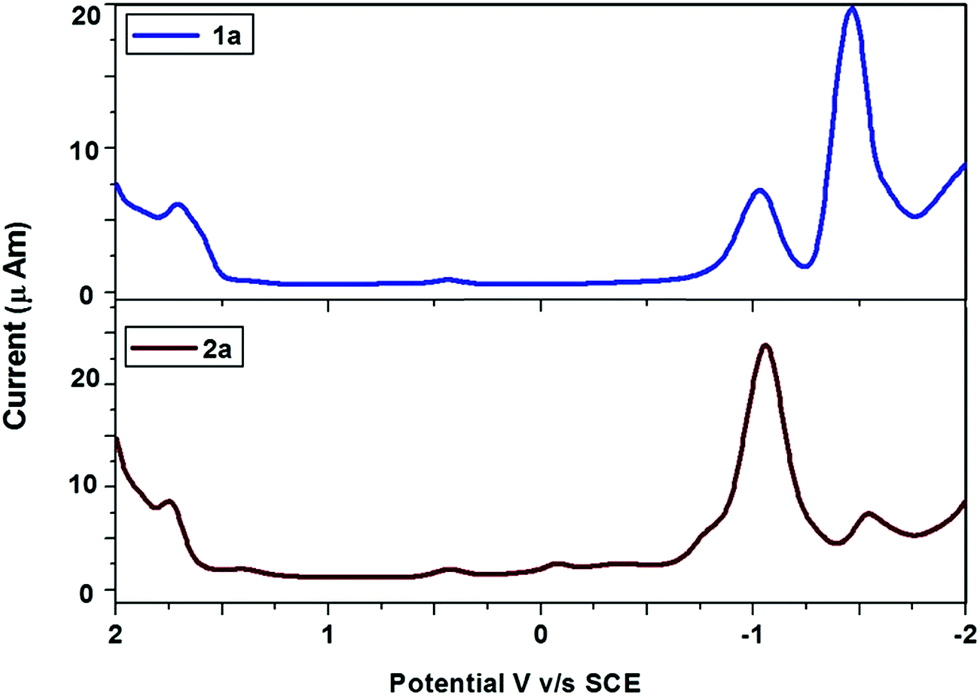 | ||
| Fig. 4 Differential pulse voltammogram of ligand 1a and platinacycle 2a. | ||
The redox potentials of the ligand and its complex is indistinguishable and difficult to assign. Therefore, we have adopted a spectroelectrochemical technique to distinguish. Using spectroelectrochemical technique, we could study both electrochemically reversible reduction and oxidation of organo platinum complexes.
Fig. 5 shows the spectral changes of Pt(IV)-complex 2a at controlled potential electrolysis. During the controlled potential reduction at −1.3 V, the intensity of absorption maxima at 235 nm increases while there was decrease in the absorption of the shoulder at 266 nm. The peak almost disappeared at 300 nm. The long wavelength MLCT band started decreasing its intensity at 350 nm. During this process, the isosbestic points were observed at 220, 250, 305 and 445 nm. The reduction was probably due to the conversion of Pt(IV) to Pt(III). When applied potential changes to −1.60 V, no changes were observed. Therefore the two reductions appeared at −1.03 and −1.47 V belong to both ligand and metal centered in complex 2a.
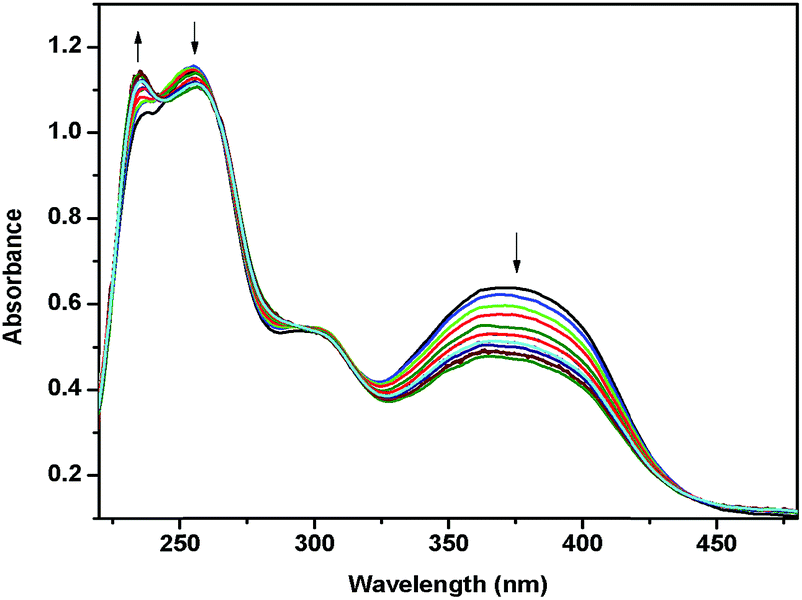 | ||
| Fig. 5 In situ UV-visible absorption changes of Pt-complex 2a at an applied potential of −1.30 V. | ||
When applied potential was at 1.80 V, the longer wavelength MLCT band at 350 nm started decreasing in intensity as shown in Fig. 6. In contrast, the intraligand π–π* transitions at 300 nm started increasing. The other intraligand absorption at 266 remained unchanged. During this process, clear isosbestic points were observed at 250 and 345 nm. As the oxidation of Pt(IV) to Pt(V) is not possible, the oxidations are probable and belong to ligand centered. Similar spectral changes were observed in other complexes during applied potential (see ESI†).
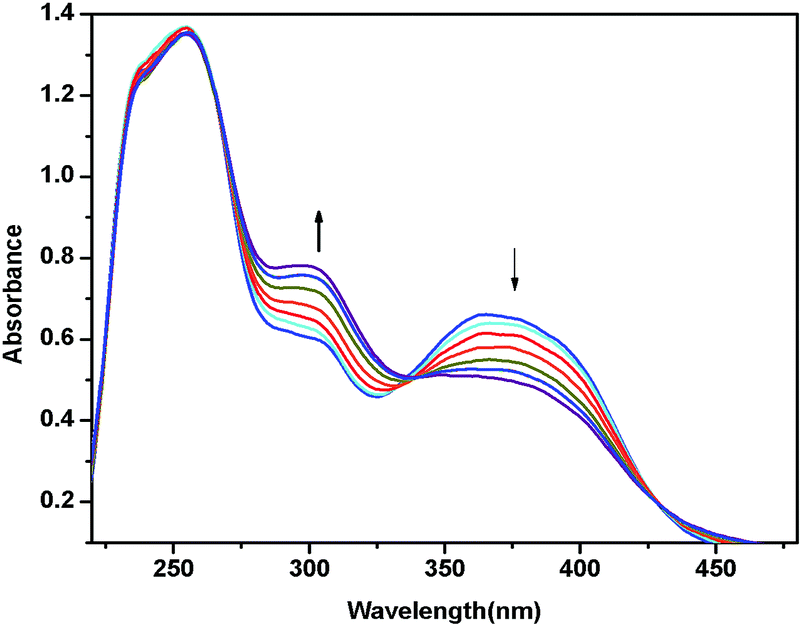 | ||
| Fig. 6 In situ UV-visible absorption changes of Pt-complex 2a at an applied potential of 1.80 V. | ||
Emission study of platinacycles 2
We have carried out steady-state emission study of both ligands and Pt(IV) complexes in dichloromethane solvent at room temperature. We have collected the emission spectra of ligands by choosing the excitation wavelength at 270 nm as shown in Fig. 7. In all the four ligands, we have observed three emission peaks at 310, 353 and 372 nm with a stoke shift of 40 nm (Table 1). Fig. 8 demonstrates irradiation low-energy band (MLCT) of all Pt(IV) complexes at ambient temperature in dichloromethane solution. All the four complexes have shown emission maxima at around 425 nm with stoke shifts of 55–65 nm.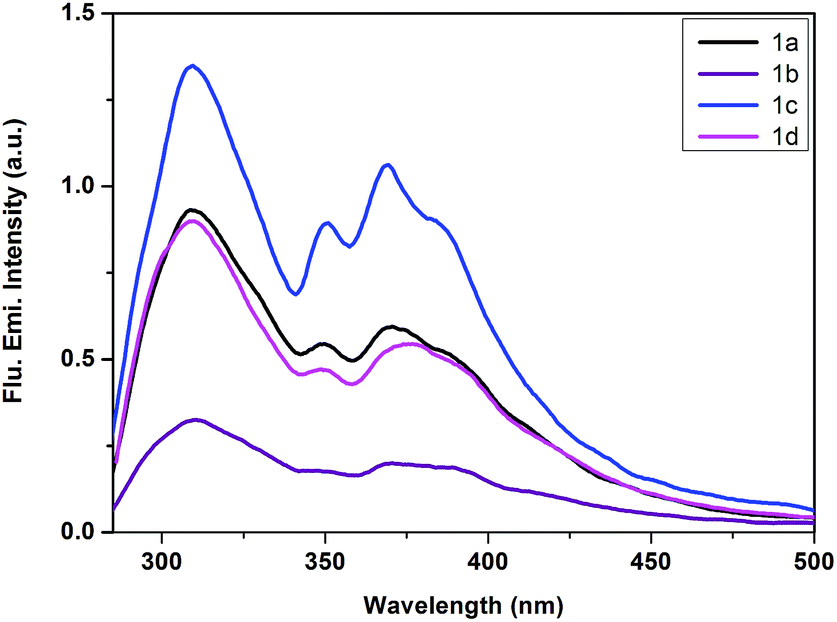 | ||
| Fig. 7 Normalised emission spectra of ligands 1a, 1b, 1c and 1d in degassed CH2Cl2 solution (2.4 × 10−4 M) at 298 K. | ||
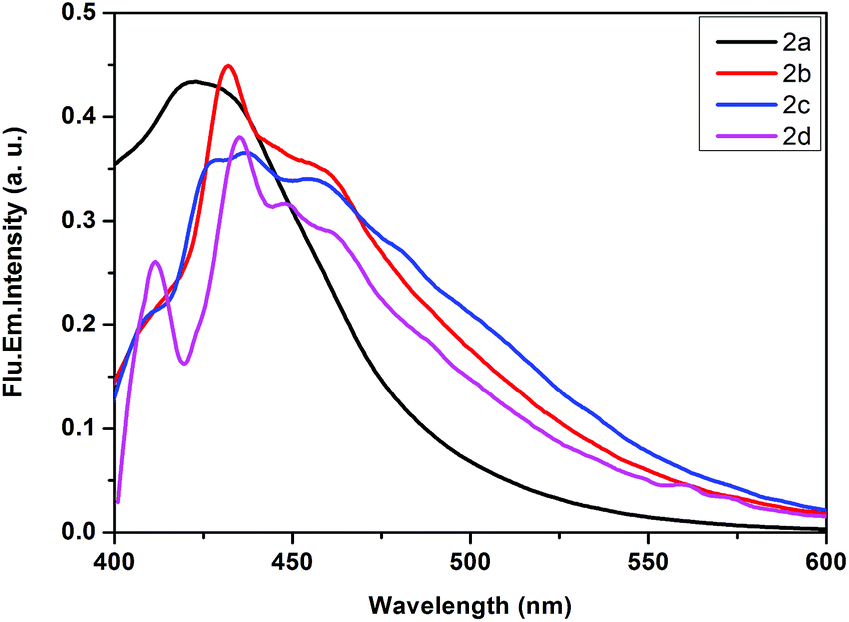 | ||
| Fig. 8 Normalised emission spectra of platinacycles 2a, 2b, 2c and 2d in degassed CH2Cl2 solution (2.4 × 10−4 M) at 298 K. | ||
Reactivity of platinacycles 2
The insertion reactions of alkynes and carbon mono oxide have been extensively studied with dimeric palladacycles and platinacycles with the formation of expanded ring system.19 Here we investigated the reactivity of Pt(IV)-complex 2b in the insertion of diphenyl acetylene (Scheme 3). The mixture of complex 2b and excess of diphenyl acetylene in dry toluene was refluxed for 12 h under nitrogen atmosphere to afford benzo[4,5] thieno[2,3c] pyridine skeletal in quantitative yield.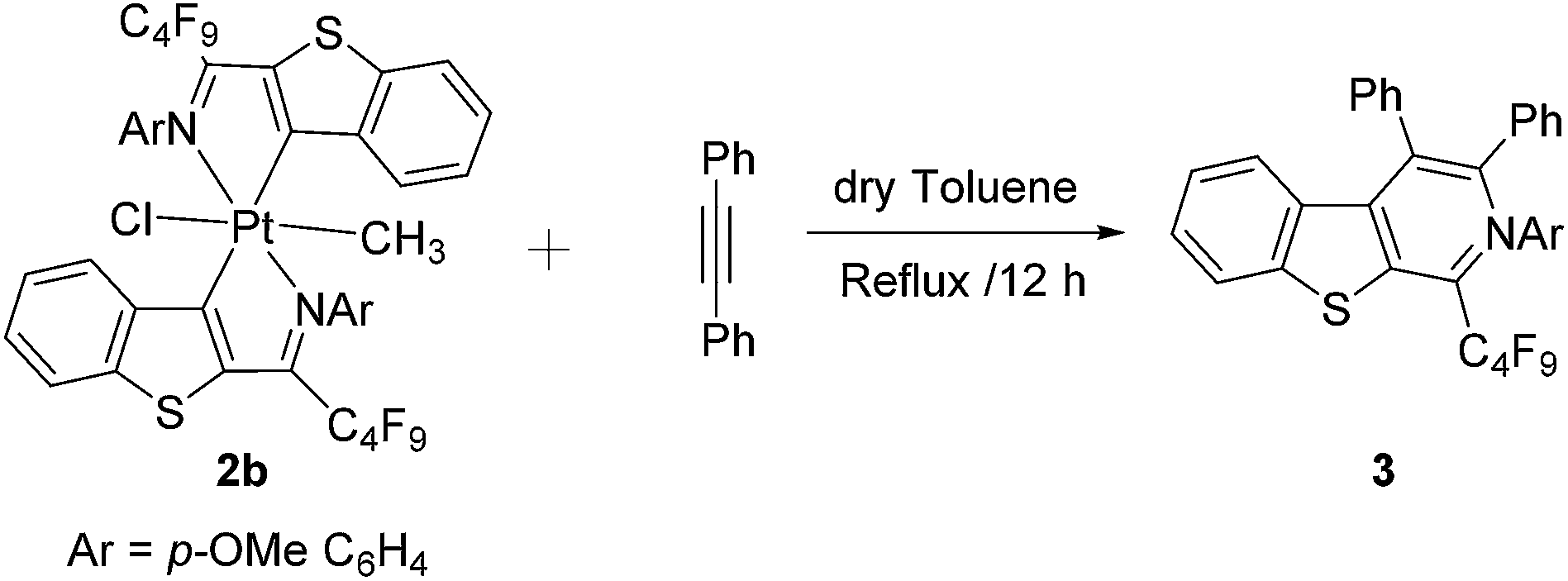 | ||
| Scheme 3 Reactivity of platinacycle 2b with diphenyl acetylene. | ||
The reactivity of Pt(IV)-complex 2a was further screened in the reaction with triphenyl phosphine (Scheme 4). Interestingly, platinacycle(IV) complex 2a was reduced to Pt(II) under the treatment of excess triphenyl phosphine in dry CDCl3 at room temperature and formed monoligated Pt(II) complex quantitatively.20
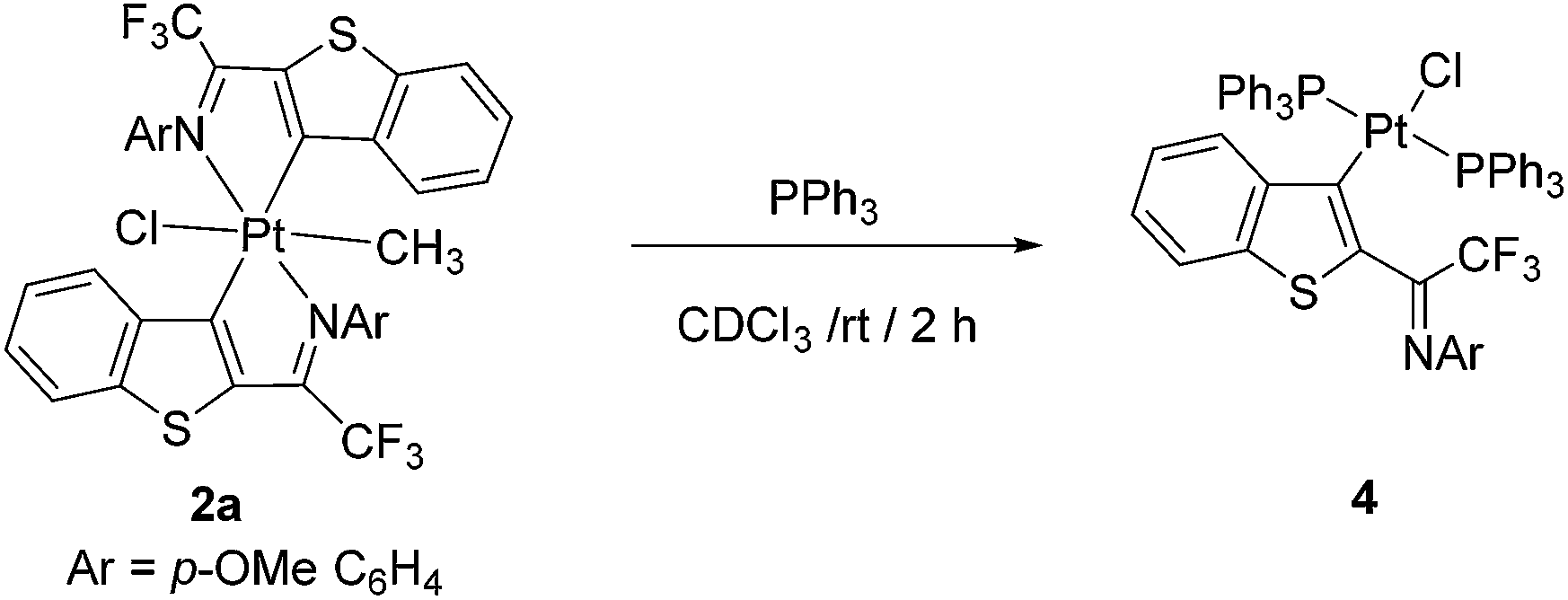 | ||
| Scheme 4 Reactivity of platinacycle 2a with triphenyl phosphine. | ||
Experimental
All reactions were carried out in oven dried glassware and under an atmosphere of nitrogen using Schlenk line. The chemicals were purchased from Aldrich and used as it is unless mentioned otherwise. All the solvents used for the reaction were dried before use. The purification of product was accomplished by column chromatography using silica gel 60–120 mesh. The technical grade solvents were used for chromatography and distilled prior to use. NMR spectra were recorded in Fourier transform mode. The 1H NMR and 13C NMR spectra were recorded on a Bruker-Avance (300 MHz); Inova (400 MHz) and Avance (500 MHz) spectrophotometer using CDCl3 and TMS as the internal standard. Multiplicities in the 1H NMR spectra are described as: s = singlet, d = doublet, t = triplet, q = quartet, qt = quintet, m = multiplet, bs = broad singlet; coupling constants are reported in Hz. Infrared spectra were recorded on Thermo Nicolet Nexus 670 spectrometer and reported in cm−1. The elemental (CHN) analysis was performed in vario MICRO cube. The perfluoroalkyl imidoyl iodides and alkynes were prepared according to literature procedure. The electrochemical measurements were performed on a PC-controlled CH instruments model CHI 620C electrochemical analyzer. The experiments were performed on 1 mM solution of either ligand or complex in CH2Cl2 solvent at scan rate of 100 mV s−1 using 0.1 M tetrabutyl ammonium perchlorate (TBAP) as supporting electrolyte. The working electrode is glassy carbon, standard calomel electrode (SCE) is the reference electrode and platinum wire is an auxiliary electrode. After a cyclic voltammogram (CV) had been recorded, ferrocene was added, and a second voltammogram was measured. The optical thin layer electrochemical studies were carried on Maya 2000 Ocean Optics software using DT-MINI-2-GS, UV-VIS-NIR LIGHTSOURCE. Steady-state fluorescence spectra was recorded using a Fluorolog-3 spectrofluorometer (Spex model, Jobin Yvon) for solutions with optical density at the wavelength of excitation (λex) ≈ 0.05. In situ ESI-MS analysis was performed using Exactive Orbitrap mass spectrometer in positive ion mode and its mass resolution was 50000 (FWHM). The sample was diluted with three volumes of acetonitrile and the resulting solution (10 μL) was injected into the source of mass spectrometer using flow-injection mode (mobile phase, methanol at a flow rate of 0.4 mL min−1).
General procedure for the preparation of N-aryl perfluoroalkyl bis(methylthio)phenyl propargyl imines (1a, 1b, 1c, 1d)
To a stirred mixture of PdCl2(PPh3)2 (2 mol%) and CuI (4 mol%) in Et3N (4 mL), 2-(methylthio/methylseleno)phenylacetylene (1 mmol) and N-aryl trifluoromethylimidoyl iodide (1 mmol) were added successively under N2 atmosphere. The mixture was stirred at room temperature until the starting materials were consumed. The reaction mixture was then filtered and from the filtrate the solvent was evaporated under reduced pressure. The crude product obtained was purified by column chromatography using hexane/EtOAc (90:10) mixture.
General procedure for the preparation of platinacylces (2a, 2b, 2c, 2d)
To a solution of PtCl2 (67 mg, 0.25 mol) in dry toluene (5 mL) at 0 °C, ligand 1 (0.25 mol) was added under N2 atmosphere. The mixture was stirred for overnight at room temperature. On the completion of reaction (monitored by TLC), the mixture was concentrated to half volume. The addition of n-hexane to mixture affords dark reddish colour precipitation. The mixture was filtered and solid residue was washed with diethyl ether (10 mL). The crude product was subjected to column chromatography and purified using hexane/EtOAc (80:20) mixture.
−237]+.599 reflections collected, 6105 unique (Rint = 0.0397). Final GooF = 1.018, R1 = 0.0293, wR2 = 0.0735, R indices based on 5390 reflections with I > 2σ(I) (refinement on F2), 445 parameters, 0 restraints, μ = 4.306 mm−1.†Conclusions
In conclusion, we have prepared a series of new octahedral cycloplatinated methyl complexes from the reaction of ortho thioanisole and ortho selenoanisole substituted perfluoroalkyl propargyl imines and Pt(II)Cl2 via intramolecular hetero-cyclization. The oxidation of Pt(II) to octahedral Pt(IV) methyl complexes is attributed to the in situ generated methyl chloride during the intramolecular cyclization. Both the ligands and complexes have observed similar redox potentials. The reduction potentials of complex are probable due to reduction Pt(IV) to Pt(II). All the Pt(IV) complexes are emissive and the stoke shifts are found to be in the range of 55–65 nm. The reactivity of these complexes suggests that they can be easily undergo insertion and reduction reaction. Further studies on the bioactivity and catalytic applications of Pt(IV) complexes are in progress.Acknowledgements
DS and KJ are thankful to UGC and CSIR, New Delhi for their fellowships.Notes and references
- (a) J. P. Collman, L. S. Hegedus, J. R. Norton and R. A. Finke, in Principles and Applications of Organotransition Metal Chemistry, 2nd edn, 1987, pp. 279–354 Search PubMed; (b) J. K. Stille, in The Chemistry of the Metal–Carbon Bond, 1985, vol. 2, pp. 625–787 Search PubMed.
- (a) G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285 CrossRef CAS PubMed; (b) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644 CrossRef CAS PubMed; (c) J.-F. Fauvarque, F. Pfluger and M. Troupel, J. Organomet. Chem., 1981, 208, 419 CrossRef CAS; (d) C. Amatore, A. Jutand and A. Suarez, J. Am. Chem. Soc., 1993, 115, 9531 CrossRef CAS; (e) P.-Y. Chen, K.-S. Huang, C.-C. Tsai, T.-P. Wang and E.-C. Wang, Org. Lett., 2012, 14, 4930 CrossRef CAS PubMed.
- (a) L. T. Pilarski, N. Selander, D. Bose and K. J. Szabo, Org. Lett., 2009, 11, 5518 CrossRef CAS PubMed; (b) R. V. Belzen, C. J. Elsevier, A. Dedieu, N. Veldman and A. L. Spek, Organometallics, 2003, 22, 722 CrossRef; (c) J. Aydin, J. M. Larsson, N. Selander and K. J. Szabo, Org. Lett., 2009, 11, 2852 CrossRef CAS PubMed; (d) M. P. Lanci, M. S. Remy, W. Kaminsky, J. M. Mayer and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 15618 CrossRef CAS PubMed.
- P. Sehnal, R. J. K. Taylor and I. J. S. Fairlamb, Chem. Rev., 2010, 110, 824 CrossRef CAS PubMed.
- G. K. Anderson, in Comprehensive Organometallic Chemistry II, 1995, vol. 9, pp. 431–53 Search PubMed.
- L. M. Rendina and R. J. Puddephatt, Chem. Rev., 1997, 97, 1735 CrossRef CAS PubMed.
- (a) R. V. Asselt, E. Rijnberg and C. J. Elsevier, Organometallics, 1994, 13, 706 CrossRef; (b) P. K. Monaghan and R. J. Puddephatt, Organometallics, 1985, 4, 1406 CrossRef CAS; (c) G. Ferguson, P. K. Monaghan, M. Parvez and R. J. Puddephatt, Organometallics, 1985, 4, 1669 CrossRef CAS; (d) P. K. Monaghan and R. J. Puddepatt, Organometallics, 1986, 5, 439 CrossRef CAS; (e) P. K. Byers, A. J. Canty, B. W. Skelton and A. H. White, Organometallics, 1990, 9, 826 CrossRef CAS.
- (a) R. H. Hill and R. J. Puddephatt, J. Am. Chem. Soc., 1985, 107, 1218 CrossRef CAS; (b) P. K. Monaghan and R. J. Puddephatt, Organometallics, 1986, 5, 439 CrossRef CAS; (c) M. Crespo and R. J. Puddephatt, Organometallics, 1987, 6, 2548 CrossRef CAS; (d) P. K. Monaghan and R. J. Puddephatt, J. Chem. Soc., Dalton Trans., 1988, 595 RSC; (e) J. Kuyper, R. Van der Laan, F. Jeanneaus and K. Vrieze, Transition Met. Chem., 1976, 1, 199 CAS; (f) J. Kuyper, Inorg. Chem., 1977, 16, 2171 CrossRef CAS; (g) A. J. Canty, R. T. Honeyman, B. W. Skelton and A. H. White, J. Organomet. Chem., 1992, 424, 381 CrossRef CAS.
- (a) C. M. Anderson, G. Ferguson, A. J. Lough and R. J. Puddephatt, J. Chem. Soc., Chem. Commun., 1989, 18, 1297 RSC; (b) M. Crespo, M. Martinez and J. Sales, J. Chem. Soc., Chem. Commun., 1992, 882 Search PubMed; (c) C. M. Anderson, M. Crespo, G. Ferguson, A. J. Lough and R. J. Puddephatt, Organometallics, 1992, 11, 1177 CrossRef CAS; (d) S. D. Perera and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1995, 641 RSC; (e) M. Crespo, X. Solans and M. J. Font-Bardia, J. Organomet. Chem., 1996, 518, 105 CrossRef CAS.
- C. R. Baar, G. S. Hill, J. J. Vittal and R. J. Puddephatt, Organometallics, 1998, 17, 32 CrossRef CAS.
- (a) S. S. Racharlawar, D. Shankar, M. V. Karkhelikar, B. Sridhar and P. R. Likhar, J. Organomet. Chem., 2014, 757, 14 CrossRef CAS; (b) P. R. Likhar, S. M. Salian, S. Roy, M. Lakshmi Kantam, B. Sridhar, K. V. Mohan and B. Jagadeesh, Organometallics, 2009, 28, 3966 CrossRef CAS.
- S. S. Racharlawar, A. Kumar, N. Mirzadeh, S. K. Bhargava, J. Wagler and P. R. Likhar, J. Organomet. Chem., 2014, 772–773, 182 CrossRef CAS.
- (a) C. T. Bui and B. L. Flynn, J. Comb. Chem., 2006, 8, 163 CrossRef CAS PubMed; (b) K. Uneyama and H. Watanabe, Tetrahedron Lett., 1991, 32, 1459 CrossRef CAS.
- (a) R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed; (b) Y.-M. Wu, M. Zhang and Y.-Q. Li, J. Fluorine Chem., 2006, 127, 218 CrossRef CAS; (c) H.-B. Yu and W.-Y. Huang, J. Fluorine Chem., 1998, 87, 69 CrossRef CAS.
- The compound 2b was isolated by column chromatography technique repeatedly however all the attempts were failed to purify the compound.
- (a) S. Perez, C. Lopez, A. Caubet, X. Solans and M. F. Bardía, Eur. J. Inorg. Chem., 2008, 2008, 1599 CrossRef; (b) C. Andersona, M. Crespo, M. F. Bardia, A. Klein and X. Solans, J. Organomet. Chem., 2000, 601, 22 CrossRef.
- L. M. Rendina and R. J. Puddephatt, Chem. Rev., 1997, 97, 1735 CrossRef CAS PubMed.
- (a) C. M. Anderson, M. A. Weinstein, J. Morris, N. Kfoury, L. Duman, T. A. Balema, A. K. Mueller, P. Scheetz, S. Ferrara, M. Chierchia and J. M. Tanski, J. Organomet. Chem., 2013, 723, 188 CrossRef CAS; (b) J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055 CrossRef CAS PubMed; (c) V. N. Kozhevnikov, M. C. Durrant and J. A. G. Williams, Inorg. Chem., 2011, 50, 6304 CrossRef CAS PubMed; (d) N. Chaudhury and R. J. Puddephatt, J. Organomet. Chem., 1975, 84, 105 CrossRef CAS; (e) M. Gianini, A. V. Zelewsky and H. S. Evans, Inorg. Chem., 1997, 36, 6094 CrossRef CAS PubMed; (f) C. Anderson, D. A. Freedman, M. Jennings and B. Gray, J. Organomet. Chem., 2005, 690, 168 CrossRef CAS.
- (a) L. Cuesta, D. Prat, T. Soler, R. Navarro and E. P. Urriolabeitia, Inorg. Chem., 2011, 50, 8598 CrossRef CAS PubMed; (b) R. V. Asselt, E. E. C. G. Gielens, R. E. Rulke, K. Vrieze and C. J. Elsevier, J. Am. Chem. Soc., 1994, 116, 977 CrossRef; (c) J. Chengebroyen, M. Linke, M. Robitzer, C. Sirlin and M. Pfeffer, J. Organomet. Chem., 2003, 687, 313 CrossRef CAS; (d) G. Wu, A. L. Rheingold, S. J. Geib and R. F. Heck, Organometallics, 1987, 6, 1941 CrossRef CAS.
- (a) P. T. Cheng and S. G. Nyburg, Can. J. Chem., 1972, 50, 912 CrossRef CAS; (b) B. S. Balaji, Y. Obora, D. Ohara, S. Koide and Y. Tsuji, Organometallics, 2001, 20, 5342 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General remarks, preparation of ligands, platinacycles, in situ ESI-MS study, absorption spectra, electrochemical spectra and spectroelectrochemical spectra, spectroscopic characterizations (1H NMR and 13C NMR) of the ligands and platinacycles. CCDC 1019738. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra17066k |
| This journal is © The Royal Society of Chemistry 2015 |