
One-step synthesis of α-Gal epitope and globotriose derivatives by an engineered α-galactosidase†
Abstract
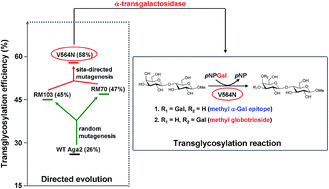
Directed evolution of an α-galactosidase (Aga2) from Bifidobacterium breve 203 by random mutagenesis and subsequently by site-directed mutagenesis provided a mutant enzyme V564N that showed high α-transgalactosylation efficiency with unobserved hydrolysis towards transglycosylation products. Using this enzyme, a one-step reaction for the simultaneous synthesis of α-Gal epitope and globotriose derivatives was achieved.
 Please wait while we load your content...
Please wait while we load your content...

