
Aqueous MW eco-friendly protocol for amino group protection†
M. Nardia,
N. Herrera Canob,
P. Costanzoc,
M. Oliverioc,
G. Sindonaa and
A. Procopioc
aDipartimento di Chimica, Università della Calabria Cubo 12C, 87036-Arcavacata di Rende (CS), Italia. E-mail: monica.nardi@unical.it
bINFIQC, Departamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Ciudad Universitaria, Córdoba 5000, Argentina
cDipartimento di Scienze della Salute, Università Magna Graecia, Viale Europa, 88100-Germaneto (CZ), Italia
First published on 9th February 2015
Abstract
In this paper a new catalyst-free and on-water method for protection of amines and amino acids with di-tert-butyl dicarbonate, 9-fluorenylmethoxycarbonyl chloride, acetyl chloride and tosyl chloride is presented. The protection can be realized in a few minutes under microwave-assistance. The reaction proved to be chemoselective in presence of ambident nucleophiles and water solution of di-tert-butyl carboxylic acid or chloride acid are the only wastes produced.
Introduction
The need to give serious consideration to the impact that human activities have on the environment has led the chemical and pharmaceutical industry to adopt the principles of green chemistry in the assessment of the degree of environmental sustainability of each synthetic process.1The essence of 12 Principles of Green Chemistry2 can be reduced to the useful working definition: Green chemistry efficiently utilizes (preferably renewable) raw materials, eliminates waste, and avoids the use of toxic and/or hazardous reagents and solvents in the manufacture and application of chemical products. Of course, it is not expected that any synthetic process will satisfy all green chemistry principles, but the more it satisfies lower its impact on the environment will be.
It has been estimated that solvents are the major contributor to the E factors of pharmaceutical manufacturing processes, amounting to about 85% of the total mass of chemicals involved in pharmaceutical processes.3 Consequently, pharmaceutical companies are focusing their effort on minimizing solvent use and in replacement of many traditional organic solvents, such as chlorinated and aromatic hydrocarbons, by more environmentally friendly alternatives including supercritical CO2, fluorous biphasic, and ionic liquids alone or in liquid–liquid biphasic combinations. Unfortunately, many of the nonconventional reaction media have prohibitive cost and/or toxicity.4
On the contrary, water appears to be a better option having many benefits: it is nontoxic, non-flammable, non-corrosive, abundantly available, and inexpensive. Moreover, the major inconveniences in using aqueous reaction mixture, such as the insolubility of most of organic reactants and the product isolation,5 can be successfully tackled by using non-conventional heating systems (microwave, ultrasound, etc.).6
Thus, in the last years the role of MW in aqueous systems has had considerable success in many organic synthesis,7,8 and protective reactions,9 although the latter are more commonly used in small-scale laboratory work and initial development than in industrial production processes because their use adds additional steps and material costs to the process. Unfortunately, the high reactivity of functional groups such as amines, alcohols and carboxylic acids, etc. makes the protection of functional groups unavoidable in multi-step organic synthesis. Thus, the protection of the amines and α-amino functionality of amino acids is one of the most important issues in synthetic organic chemistry and in peptide chemistry.10
Because most peptide syntheses, both in solution and on solid phase, are carried out in the C to N direction, α-amino protecting groups (temporary protecting groups) are used and removed several times during the synthesis, and therefore, the synthetic methods must be mild that do not affect the remaining protecting groups or even the peptidic chain.11
At this regard, sulfonamides and carbamides have become one of the most attractive protecting group for amines.12 Moreover, these derivatives are extremely useful pharmaceutical compounds for the their biological activities such anticancer,13 anti-infiammatory,14 and antiviral agents.15
In continuation of our interest towards development of useful green synthetic methodologies for the chemical of protecting groups,16 here we report a simple and ecofriendly method, the use of microwave irradiation17 for the selective synthesis of different sulfonamide and carbamide derivatives in water without the use of any catalyst.
Results and discussion
We have recently reported a MW assisted protocol for the rapid and efficient formation/cleavage of O-tert-butoxy carbonates17 using erbium(III) salts as catalysts and for the tosylation of a wide set of alcohols in the presence of anhydride derivatives.18 Now we have decided to test the potential use of MW in the protection of amines and amino acids using sulfonyl and carbonate chlorides as protecting groups.Thus, in order to investigate product distribution, preliminary experiments were carried out on the model reaction between benzylamine (1 mmol) and tosyl chloride (1.1 mmol) (Scheme 1).
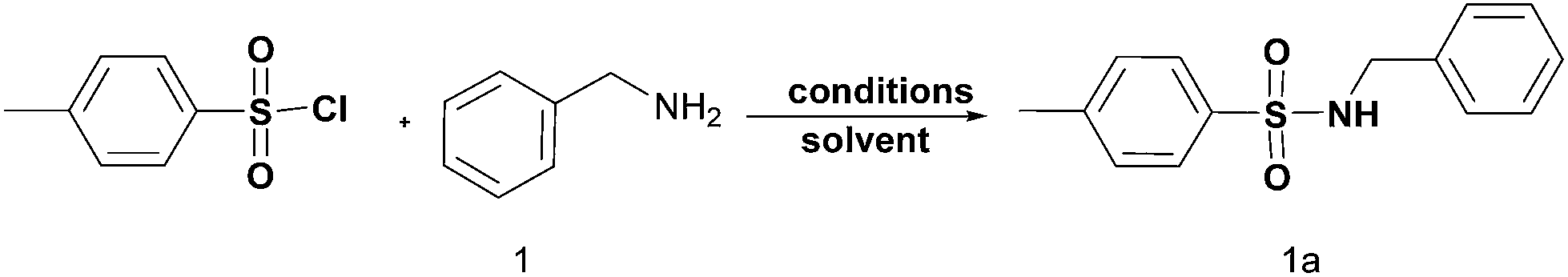 | ||
| Scheme 1 | ||
In the first instance, the reaction was performed in dichloromethane using different erbium(III) salts as catalysts. A yield of 96% is obtained only in reflux conditions regardless of the catalyst used (entries 1–5 in Table 1), while poor results were obtained when the reaction was carried out at room temperature.
| Entry | Lewis acidb | Solvent | T °C | Time min | Yieldc (%) |
|---|---|---|---|---|---|
| a General reaction conditions: 2 mmol of aniline and 2.2 mmol of tosyl chloride was dissolved in the solvent (3 mL).b 10% mol of Lewis acid.c Isolated yield.d 60 °C, 80 °C and 110 °C for entries 17, 18 and 19 respectively 20, 21 and 22 respectively.e 110 °C. | |||||
| 1 | ErCl3 × 6H2O | CH2Cl2 | r.t | 150 | 4 |
| 2 | ErCl3 × 6H2O | CH2Cl2 | 60 | 150 | 96 |
| 3 | Er(OTf)3 | CH2Cl2 | 60 | 150 | 96 |
| 4 | ErCl3 | CH2Cl2 | 60 | 150 | 96 |
| 5 | ErCl3 × 6H2O | CH2Cl2 | 80 | 150 | 97 |
| 6 | ErCl3 × 6H2O | EL | 60 | 150 | 68 |
| 7 | Er(OTf)3 | EL | 60 | 150 | 68 |
| 8 | ErCl3 | EL | 60 | 150 | 68 |
| 9 | ErCl3 × 6H2O | 2-MeTHF | 60 | 240 | 98 |
| 10 | Er(OTf)3 | 2-MeTHF | 60 | 240 | 98 |
| 11 | ErCl3 | 2-MeTHF | 60 | 240 | 98 |
| 12 | — | 2-MeTHF | 60 | 240 | 97 |
| 13 | ErCl3 × 6H2O | H2O | 60 | 150 | 94 |
| 14 | Er(OTf)3 | H2O | 60 | 150 | 98 |
| 15 | ErCl3 | H2O | 60 | 150 | 95 |
| 16 | — | H2O | 60 | 150 | 0 |
| 17d | ErCl3 × 6H2O | H2O | MW | 15 | 98 |
| 18d | Er(OTf)3 | H2O | MW | 15 | 98 |
| 19d | ErCl3 | H2O | MW | 15 | 98 |
| 20d | — | H2O | MW | 15 | 98 |
| 21d | — | H2O | MW | 10 | 98 |
| 22d | — | H2O | MW | 5 | 98 |
| 23e | — | — | MW | 17 | 90 |
Using green solvent such as ethyl lactate (EL) and 2-MeTHF the reaction yield decreases in the first case (entries 6–8 in Table 1) and increases in the second case (entries 9–11 in Table 1), but no improvement in reaction yields was observed using different catalysts of Er(III) or prolonging the reaction time in both the cases. Noteworthy, still very good result was registered when the reaction was performed in 2-MeTHF in the absence of catalyst (entry 12 in Table 1).
Lastly, the best reaction conditions were found to be using H2O as solvent, 10 mol% Er(OTf)3 as a catalyst at 60° C for 150 min (entry 14, Table 1), although no product formation was observed in the absence of catalyst (entry 16 in Table 1).
Anyhow, a marked decrease in reaction times was observed when the same reaction was performed by microwave activation at different temperatures (entries 17–19 in Table 1).
Surprisingly, still excellent results in a short reaction time were registered conducting the same MW-assisted-reaction in the absence of catalyst (entries 20–22 in Table 1). Notably, entry 23 in Table 1 clearly shows that the absence of water retards the reaction time decreasing significantly the reaction yield. This means that the water associated with the system MW plays a fundamental role in the protection reaction.19
Thus, the catalytic activity Er(III) is brilliantly replaced by the effect of MW in aqueous system and the experimental conditions depicted in Fig. 1 (see also entry 22 in Table 1) can be considered the best experimental conditions for this reaction.
 | ||
| Fig. 1 | ||
To explore the applicability of the above reported method, the same experimental protocol was applied to other protecting groups as tert-butyl carbonate (using Boc anhydride as reagent), fluorenylmethyloxycarbonyl (using FmocCl as reagent) and acetyl chloride using model substrate benzylamine (Table 2).
|
||||
|---|---|---|---|---|
| Entry | Protecting group | Condit. | Time [min] | Yieldb [%] |
| a General reaction conditions: 2 mmol of benzylamine and 2.2 mmol of Boc anhydride (entry 1) or Fmoc chloride (entries 2–4) or acetyl chloride (entry 5) was dissolved in H2O (3 mL). The reactions were conducted in a Syntos 3000 microwave oven (Anton-Paar) at different temperature.b Isolated yield. | ||||
| 1 | 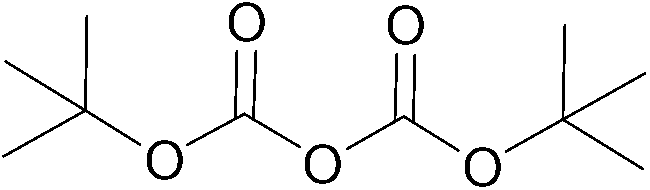 |
80 °C | 5 | 98 |
| 2 | 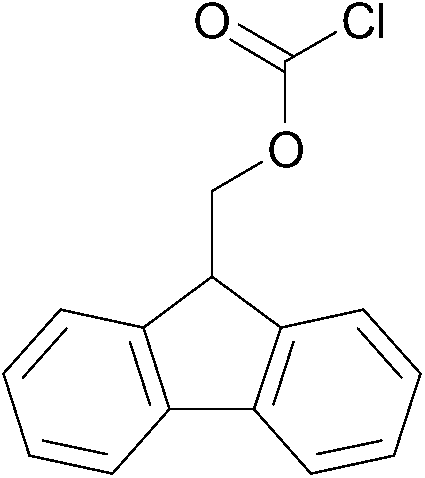 |
80 °C | 5 | 70 |
| 3 | 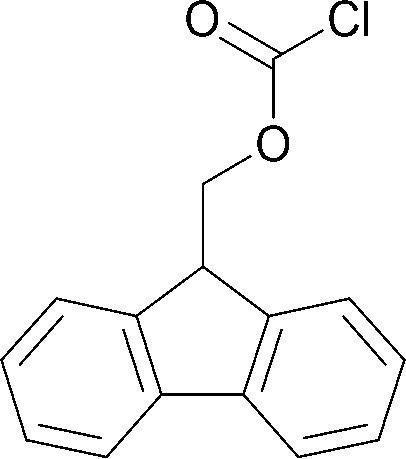 |
80 °C | 10 | 82 |
| 4 | 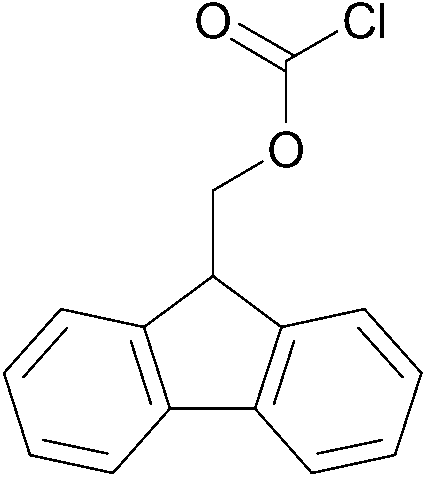 |
110 °C | 5 | 99 |
| 5 |  |
80 °C | 5 | 98 |
Quantitative yields of protected substrate were obtained for all of the three reagents in only 5 minutes (Table 2) and the need of a higher reaction temperature for the Fmoc derivative can be attributed to its high steric hindrance (entry 2 in Table 2). However, increasing the reaction times up to 10 min, 82% yield of product was obtained (entry 3 in Table 2), and quantitative yield of protected substrate was collected performing the reaction at 110 °C for 10 minutes (entry 4, Table 2).
At this point, to explore the applicability/scope of the settled method employed, we have applied the same reaction condition to different aromatic and aliphatic amines (Table 3).
| Entry | Substrate | Protecting group | Product | Yield (%) |
|---|---|---|---|---|
| a Reaction conditions: amine (2 mmol), protecting reagent (2.2 mmol), water 2 mL in MW for 5 min.b For 2b, 3b, 2c and 3c the reaction was carried out for 10 min. | ||||
| 1 |  |
TosylCl | 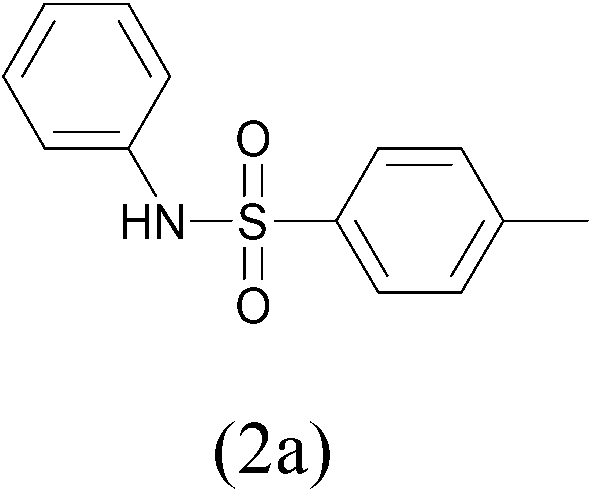 |
98 |
| FmocCl | 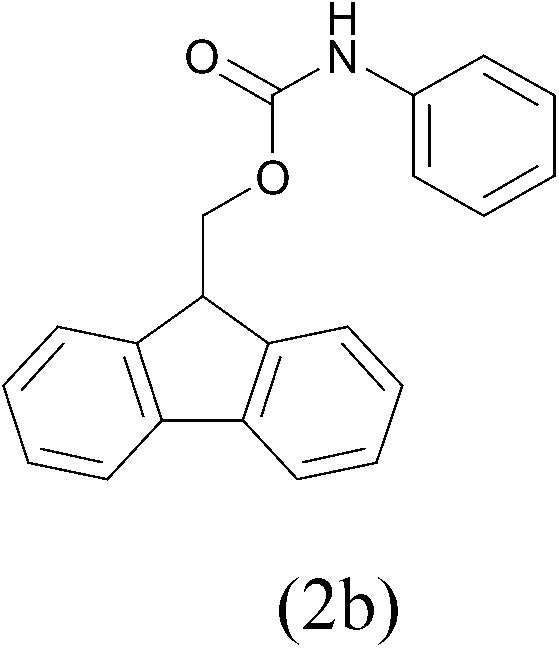 |
92b | ||
| (Boc)2O |  |
94b | ||
| AcCl | 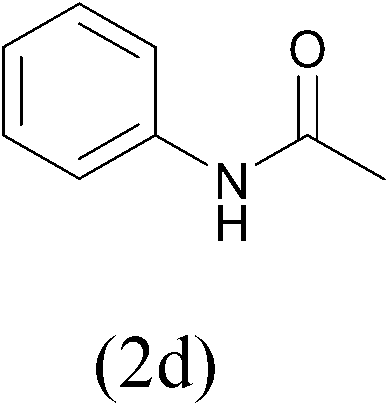 |
98 | ||
| 2 | 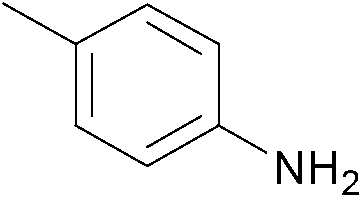 |
TosylCl | 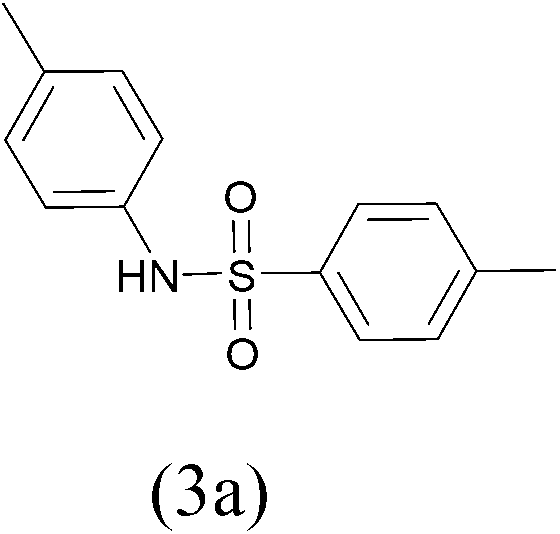 |
98 |
| FmocCl | 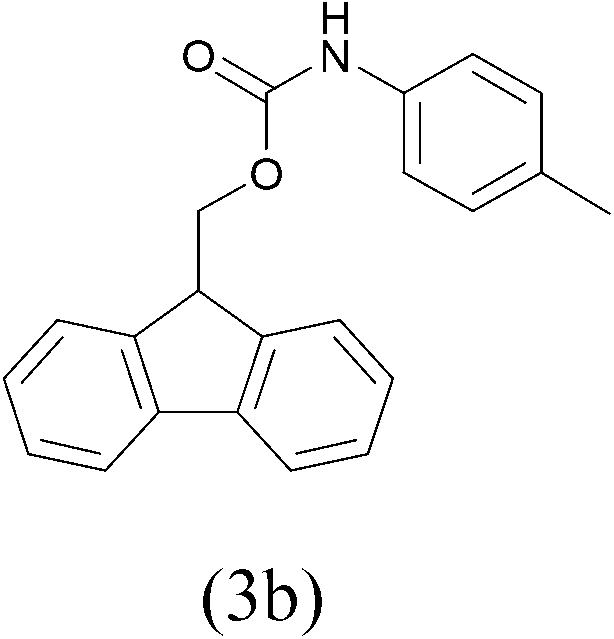 |
90b | ||
| (Boc)2O |  |
98b | ||
| AcCl | 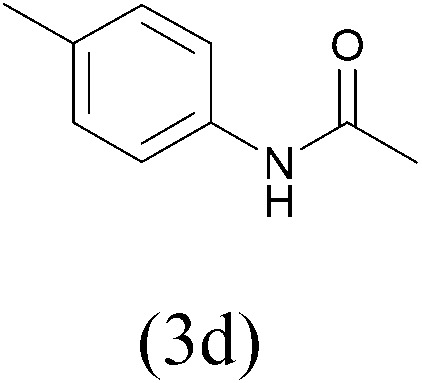 |
97 | ||
| TosylCl | 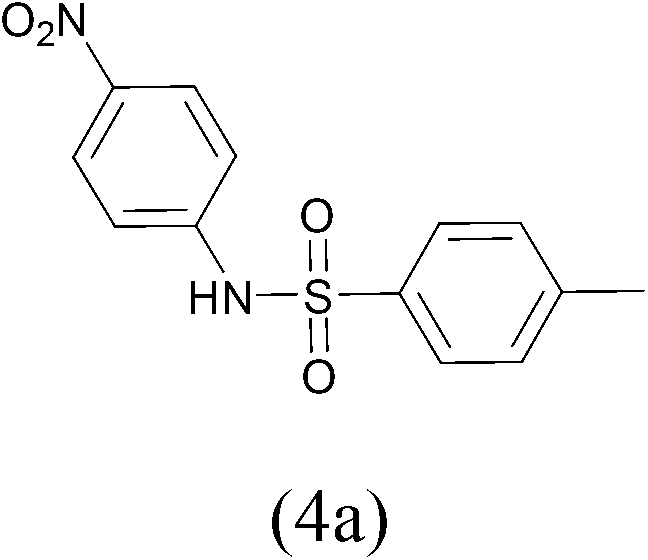 |
85 | ||
| 3 | 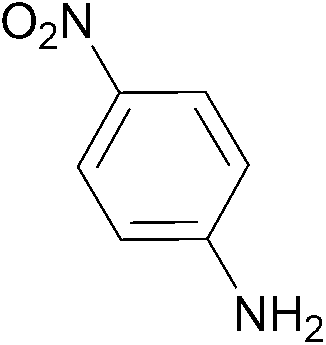 |
FmocCl | 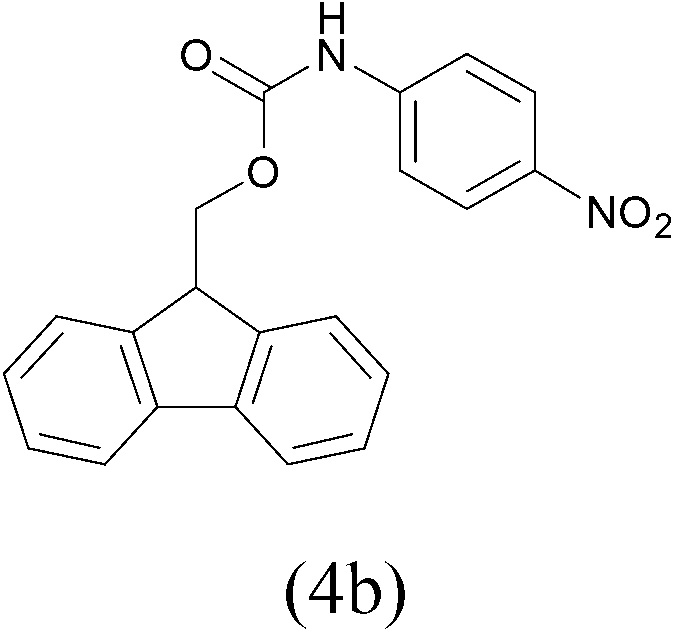 |
80 |
| (Boc)2O | 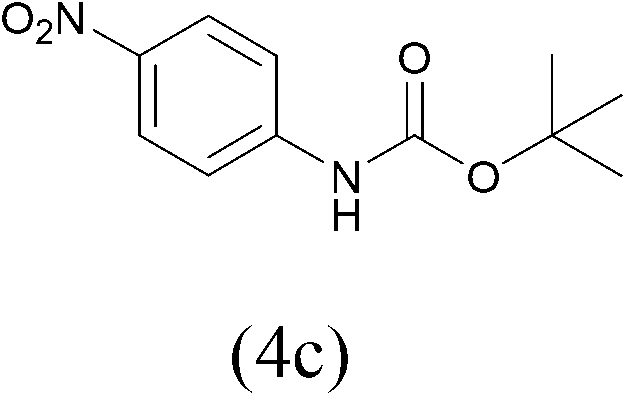 |
98 | ||
| AcCl | 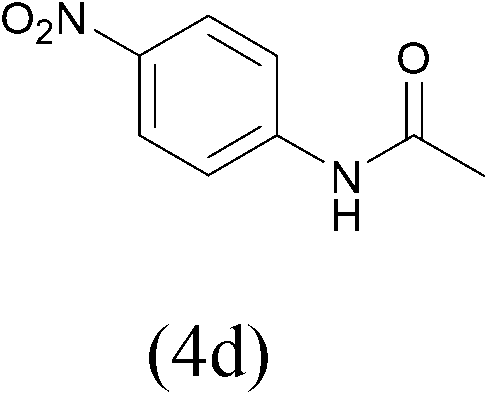 |
96 | ||
| 4 | 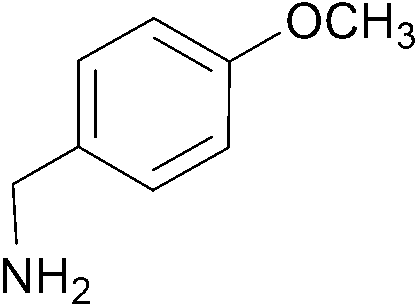 |
TosylCl | 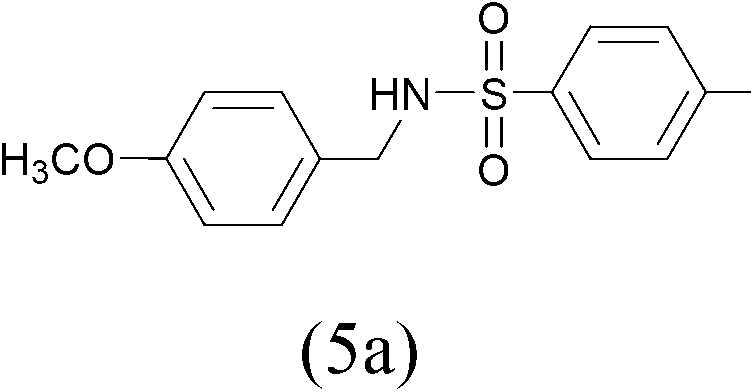 |
98 |
| FmocCl | 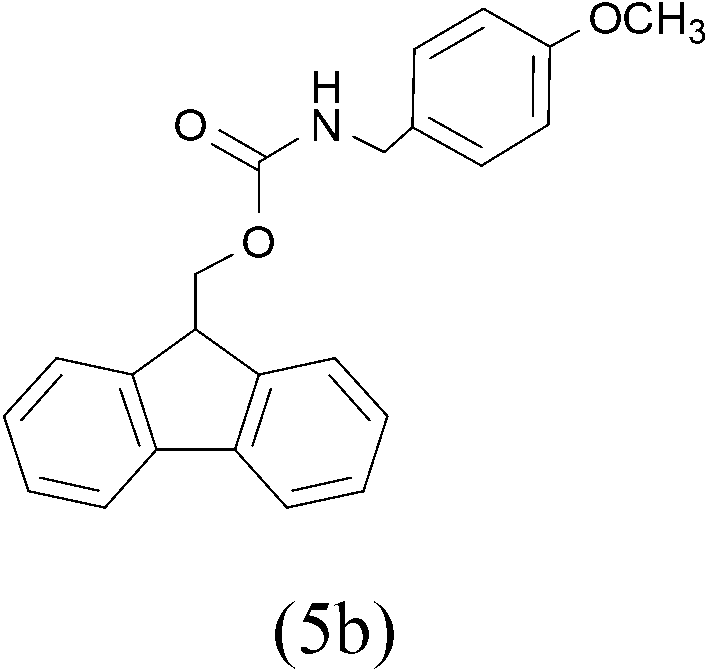 |
95 | ||
| (Boc)2O | 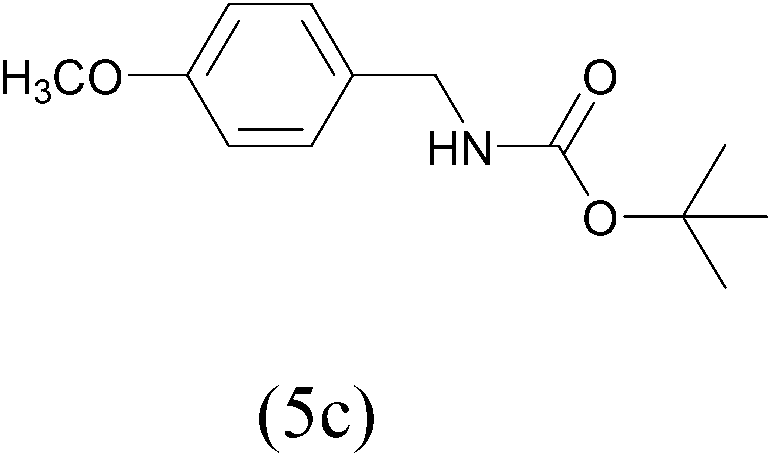 |
98 | ||
| AcCl | 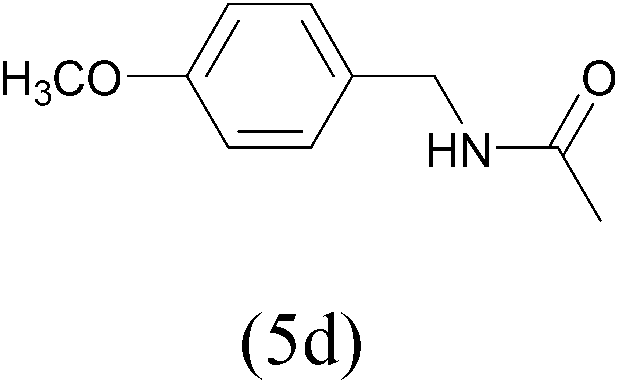 |
97 | ||
| 5 | CH3(CH2)7NH2 | TosylCl | 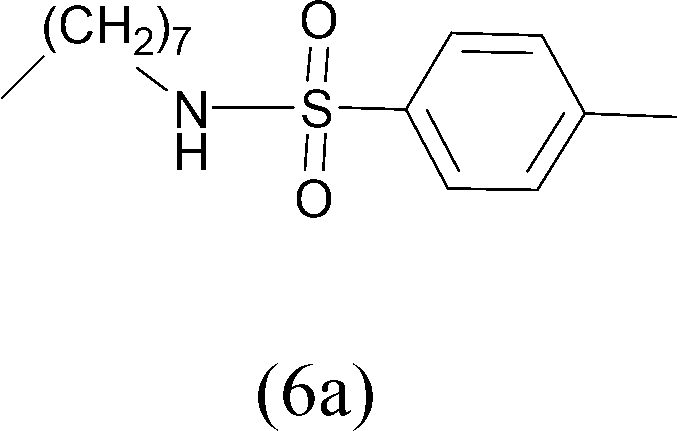 |
83 |
| FmocCl |  |
95 | ||
| (Boc)2O |  |
91 | ||
| AcCl |  |
97 | ||
All the aromatic amines gave high yield of protected substrates (entries 1–4 in Table 3) and only for the p-nitroaniline lower yield of tosyl and Fmoc derivatives were observed (4a and 4b entry 3 in Table 3); probably due to the presence of electron-withdrawing group on the aromatic ring.
Lastly, the good performances reported in Table 3 for the protection reactions conducted on various amines showed the efficiency of the presented protocol, pushing our interest to explore its applicability as rapid and efficient protection method of amino acids (Scheme 2).
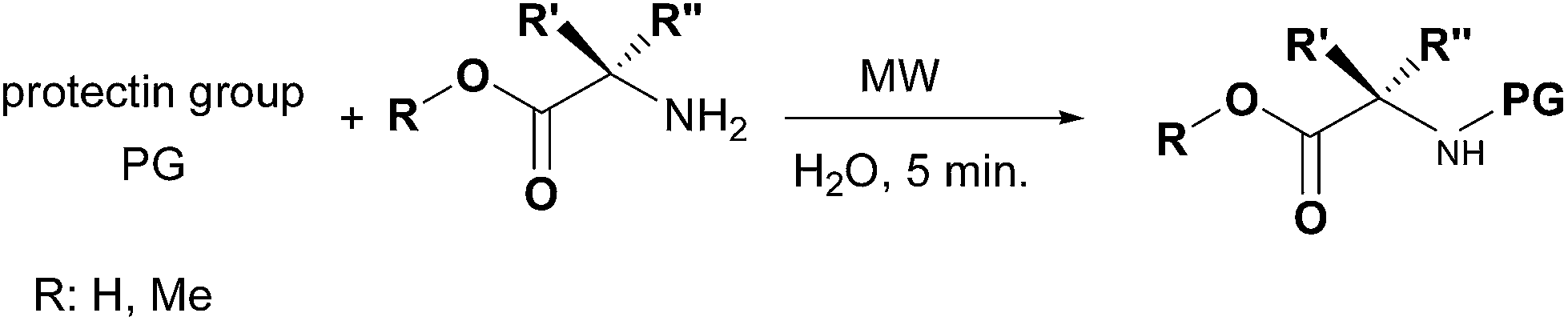 | ||
| Scheme 2 | ||
In Table 4 are described the results of the protection reactions performed on a series of free or O-methylated amino acids. In all the reported examples, the N-protected amino acids were obtained in excellent yields in only 5 minutes, therefore making this method particularly appropriate for the environmentally friendly protection and deprotection of amino acids.
| Entry | Substr. | Protecting group | Product | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: amino acid (2 mmol), protecting group (2.2 mmol), water 2 mL, MW, at 110 °C for 5 min.b Isolated yield. | ||||
| 1 | 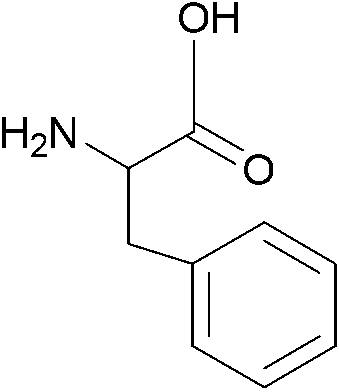 |
TosylCl | 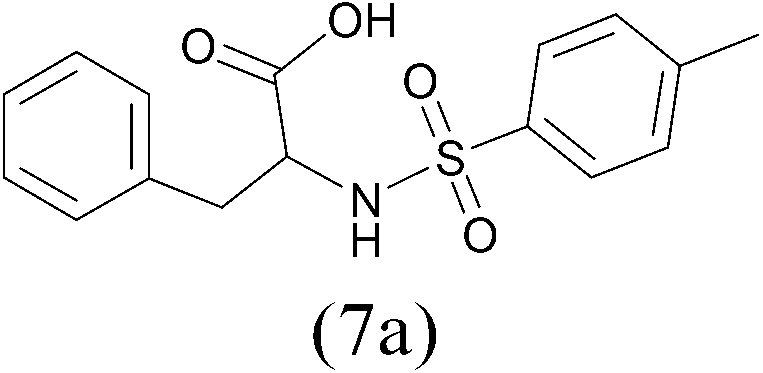 |
98 |
| FmocCl | 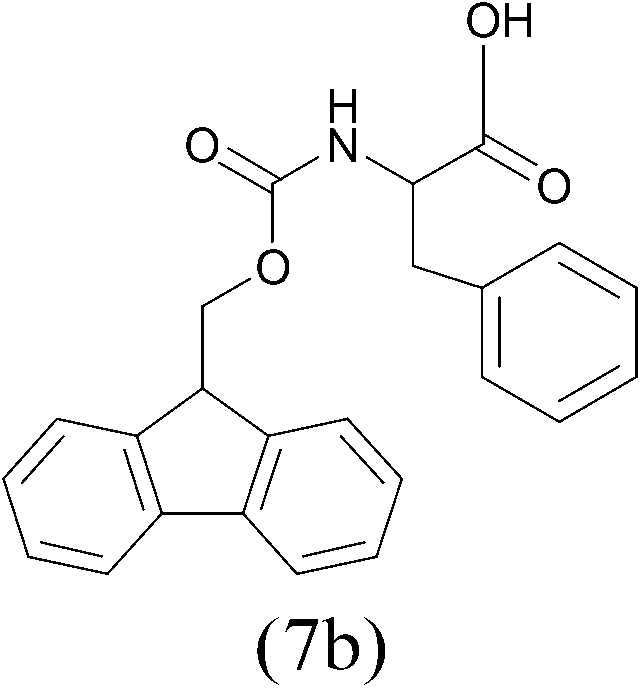 |
92 | ||
| (Boc)2O | 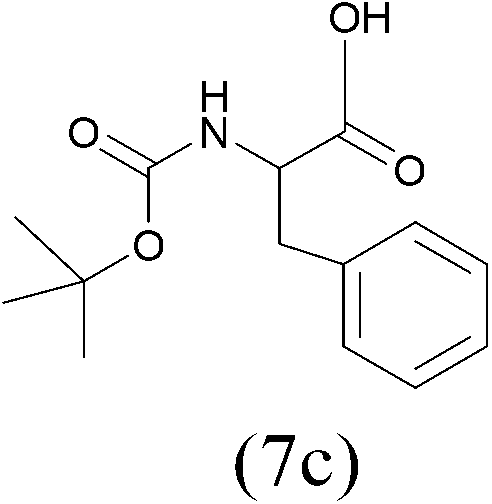 |
94 | ||
| AcCl | 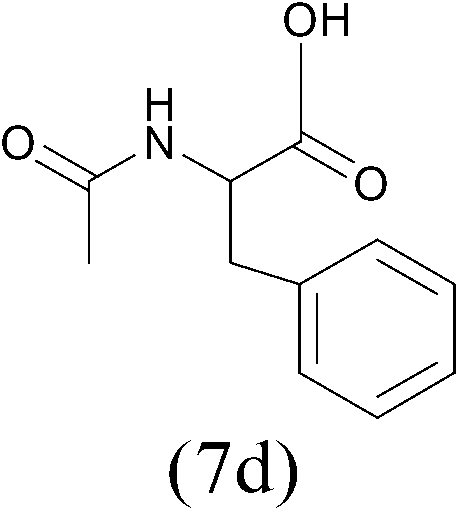 |
97 | ||
| 2 | 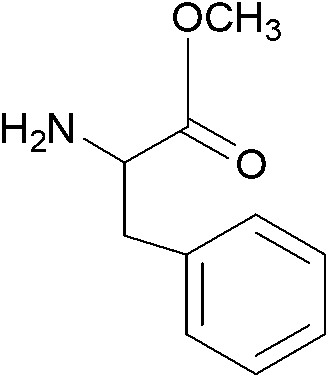 |
TosylCl |  |
94 |
| FmocCl | 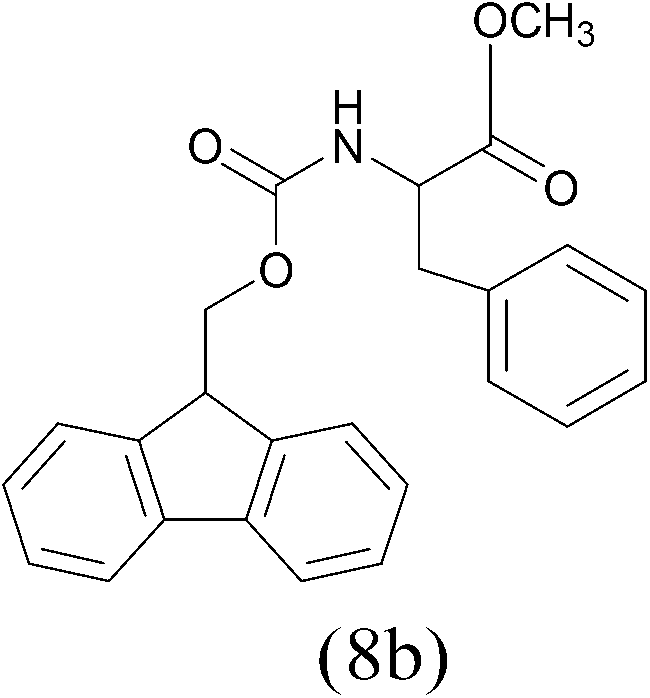 |
92 | ||
| (Boc)2O | 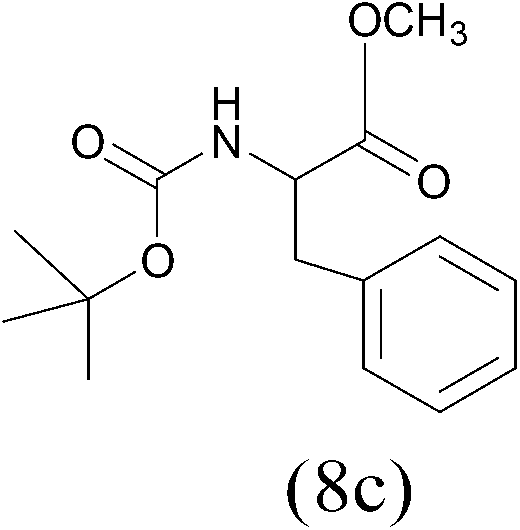 |
96 | ||
| AcCl | 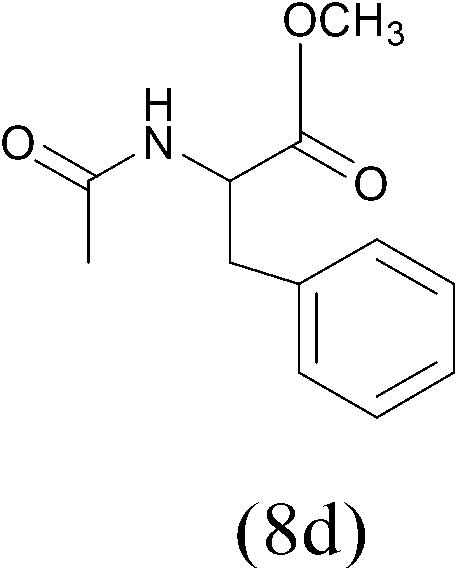 |
95 | ||
| 3 |  |
TosylCl | 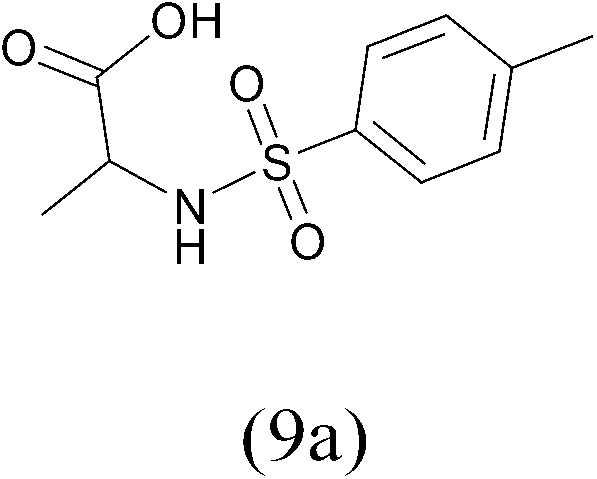 |
96 |
| FmocCl | 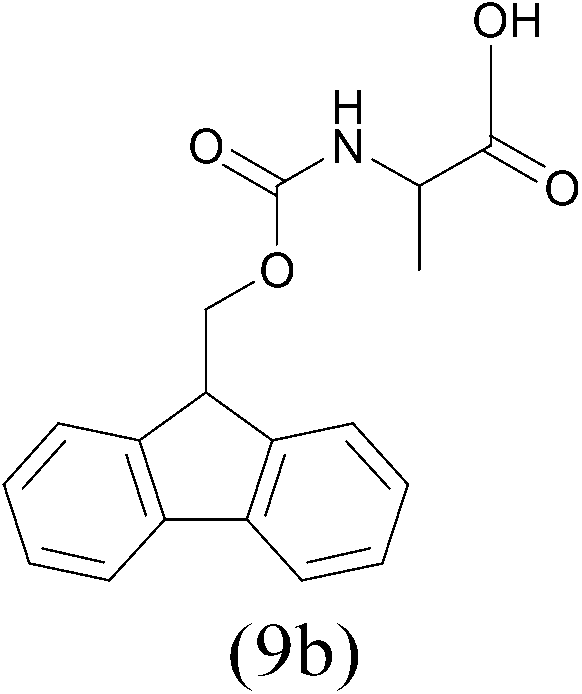 |
96 | ||
| (Boc)2O | 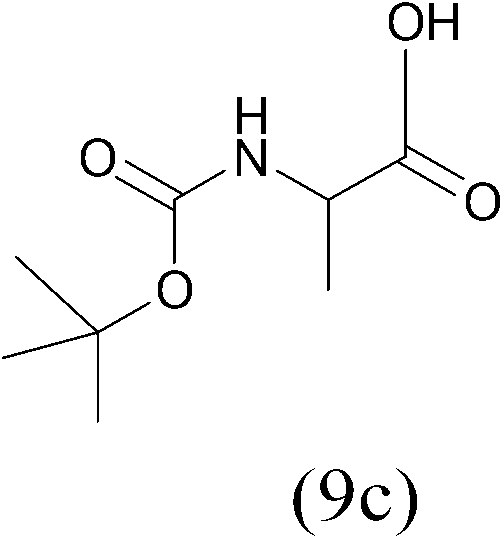 |
80 | ||
| AcCl | 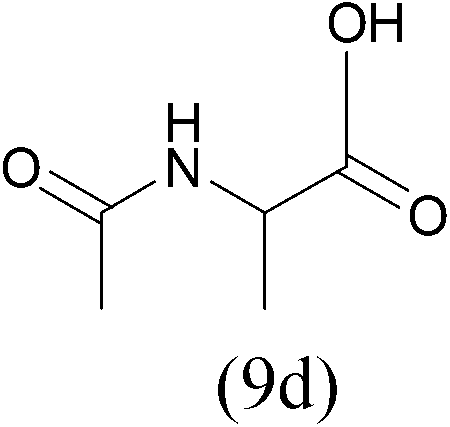 |
85 | ||
| 4 | 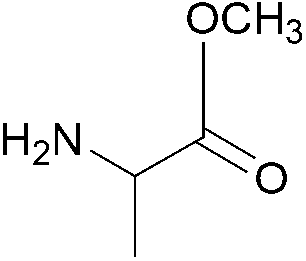 |
TosylCl | 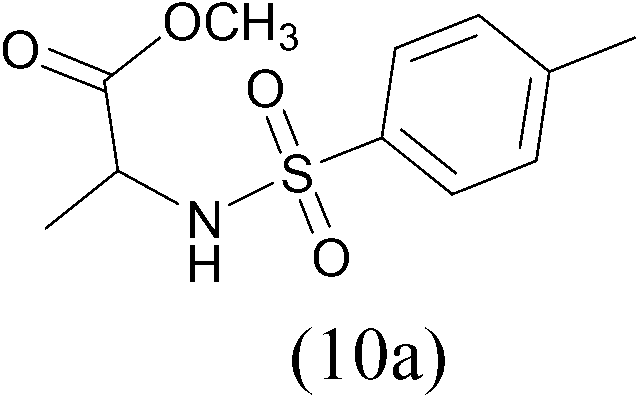 |
98 |
| FmocCl | 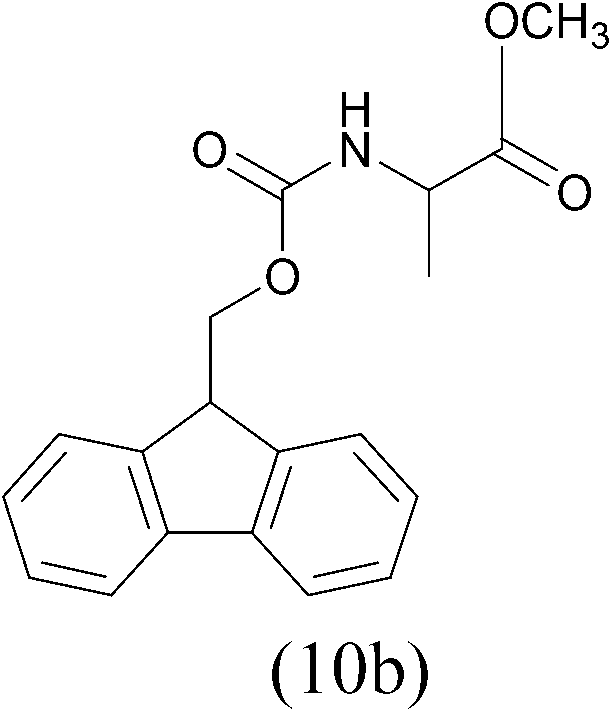 |
92 | ||
| (Boc)2O | 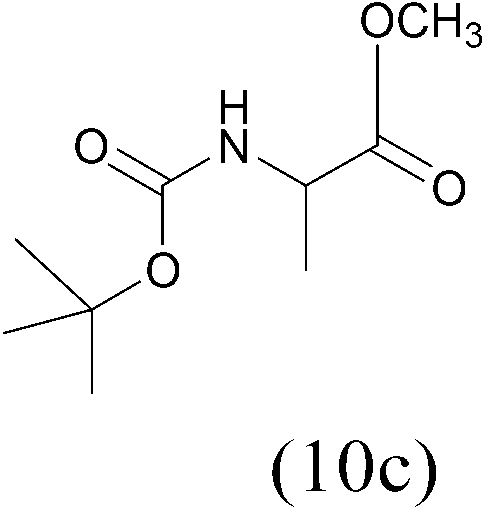 |
96 | ||
| AcCl | 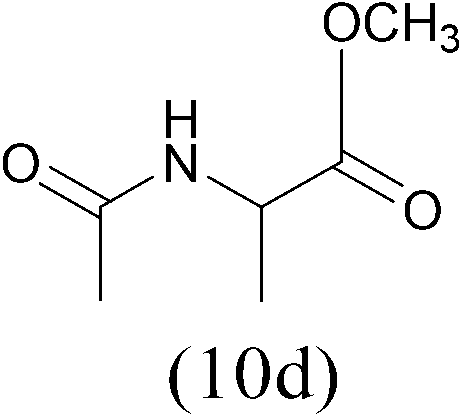 |
97 | ||
| 5 | 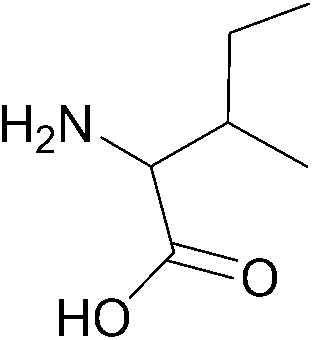 |
TosylCl | 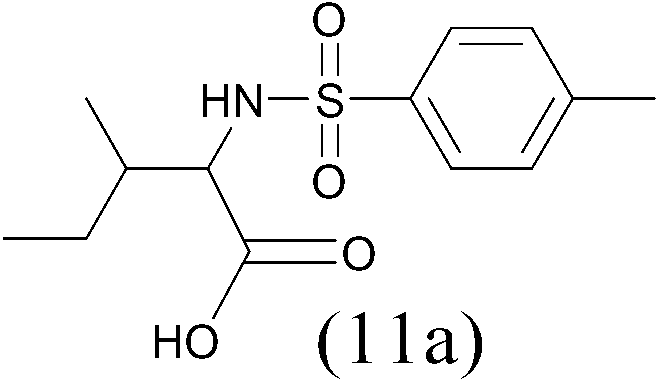 |
98 |
| FmocCl | 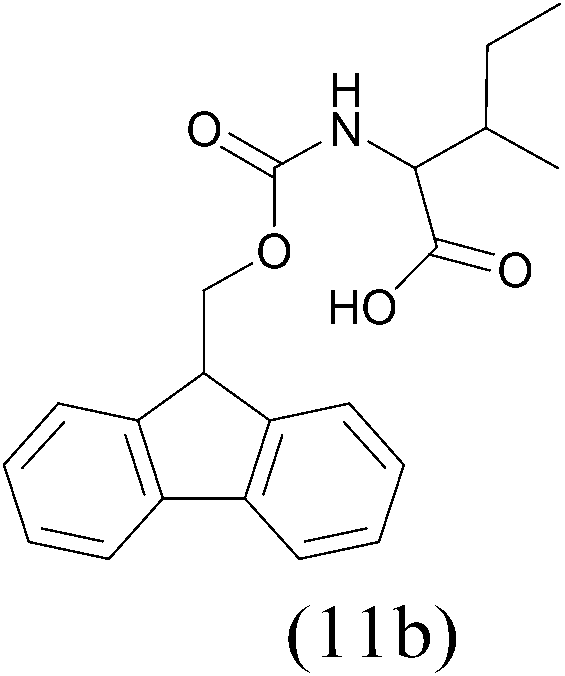 |
80 | ||
| (Boc)2O | 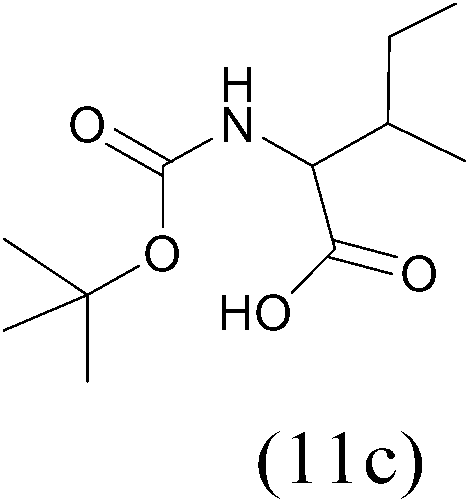 |
85 | ||
| AcCl | 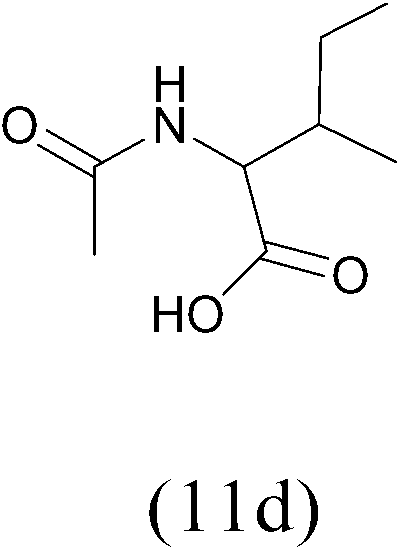 |
87 | ||
| 6 | 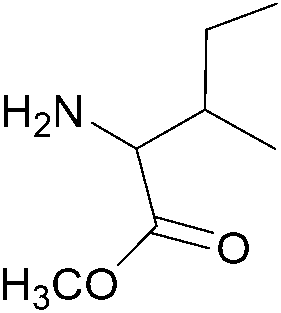 |
TosylCl |  |
97 |
| FmocCl | 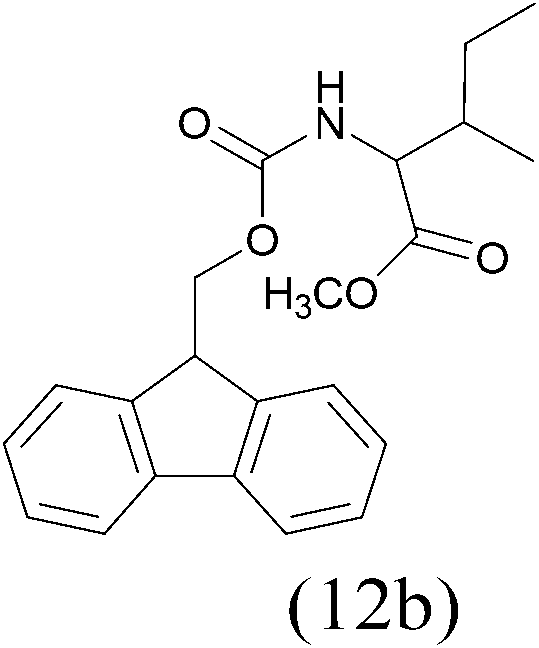 |
93 | ||
| (Boc)2O | 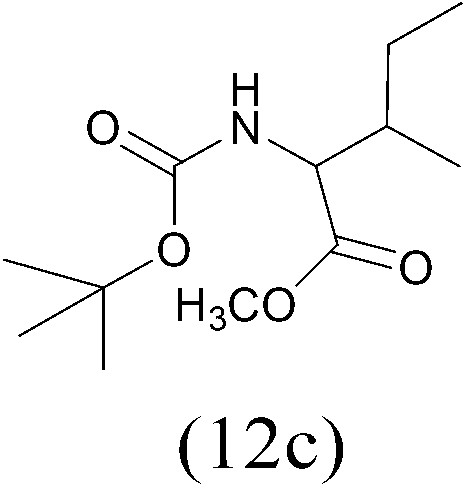 |
97 | ||
| AcCl |  |
95 | ||
Experimental section
General
All chemicals and solvents were purchased from common commercial sources and were used as received without any further purification. All reactions were monitored by TLC on silica Merck 60 F254 percolated aluminum plates and were developed by spraying with ninhydrin solution when possible or with a GC-MS Shimadzu workstation, constituted by a GC 2010 (provided of a 30 m-QUADREX 007-5MS capillary column, operating in “splitless” mode, 1 ml min−1 flow of He as carrier gas) and a 2010 quadrupole mass-detector. All reactions were carried out in the a Syntos 3000 Microwave oven (Anton-Paar) with rotor 4 × 24MG5. Proton nuclear magnetic resonance (1H-NMR) spectra were recorded on a Brücker spectrometer at 300 MHz. Chemical shifts are reported in δ units (ppm) with TMS as reference (δ 0.00). All coupling constants (J) are reported in Hertz. Multiplicity is indicated by one or more of the following: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet). Carbon nuclear magnetic resonance (13C-NMR) spectra were recorded on a Brücker at 75 MHz. Chemical shifts are reported in δ units (ppm) relative to CDCl3 (δ 77.0). Melting points were obtained on a Kofler apparatus.General procedure to determine the substrate scope
To a stirred solution of tosyl chloride Fmoc chloride, Boc anhydride (2.2 mmol), in H2O (3 mL) was added the N-nucleophile (2.0 mmol) and conducted in MW. The reaction mixture until TLC indicated consumption of the starting material. The reaction mixture presents a organic precipitate The same, repeatedly washed with water is subsequently dried.Conclusions
Synthetic organic protocols using greener and more efficient methodologies are crucial for the development of more sustainable processes. This article highlights the importance of water in organic synthesis and exhibits a novel and efficient route for water-mediated N-protection of amines with MW. The absence of acid/base and the use of water makes present procedure environmentally friendly. In the light of these premises, further developments of benign by design methodologies are expected in the coming study. The authors believe that these methods of sustainable protection are stimulus to the development of synthetic methods for peptides that meet the 12 principles of Green Chemistry.2Notes and references
- (a) P. J. Dunn, A. S. Wells and M. T. Williams, Green Chemistry in the Pharmaceutical Industry, Wiley-VCH Verlag GmbH, Weinheim, 2010, ISBN: 978-3-527-32418-7 Search PubMed; (b) M. B. Gawande, V. D. B. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522 RSC; (c) M. B. Gawande and P. S. Branco, ChemSusChem, 2014, 7, 24 CrossRef CAS PubMed.
- (a) Green Chemistry: Theory and Practice, ed. Anastas, P. T., and Warner, J. C., Oxford University Press, Oxford, 1998 Search PubMed; (b) P. T. Anastas and J. C. Warner, Green Chemistry: Frontiers in Chemical Synthesis and Processes, Oxford Science Publications, Oxford, 1998 Search PubMed.
- C. Jiminez-Gonzales, A. D. Curzons, D. J. C. Constable and V. L. Cunningham, Int. J. Life Cycle Assess, 2004, 9, 115–121 Search PubMed.
- Y. Zhang, B. R. Bakshi and E. Sahledemessie, Environ. Sci. Technol., 2008, 42, 1724 CrossRef CAS.
- (a) C. J. Li and L. Chen, Chem. Soc. Rev., 2006, 35, 68 RSC; (b) A. De Nino, L. Maiuolo, P. Merinos, M. Nardi, A. Procopio and D. Roca-López, ChemCatChem, 2015 DOI:10.1002/cctc.201402973.
- (a) J. H. Clark, Green Chem., 2006, 8, 17 RSC; (b) C. J. Li and L. Chen, Chem. Soc. Rev., 2006, 35, 68 RSC; (c) V. Polshettiwar and R. S. Varma, Chem. Soc. Rev., 2008, 37, 1546 RSC; (d) D. Dallinger and C. O. Kappe, Chem. Rev., 2007, 107, 2563 CrossRef CAS PubMed; (e) L. Leseurre, J.-P. Genet and V. Michelet, in Green Solvents, Handbook of Green Chemistry, ed. P. T. Anastas and C.-J. Li, Wiley-VCH Verlag GmbH, Weinheim, 2009 Search PubMed; (f) V. Polshettiwar and R. S. Varma, Chem. Soc. Rev., 2008, 37, 1546–1557 RSC.
- (a) C. R. Strauss and R. W. Trainor, Aust. J. Chem., 1995, 48, 1665 CrossRef CAS; (b) C. R. Strauss and R. S. Varma, Top. Curr. Chem., 2006, 266, 199 CrossRef CAS; (c) A. K. Rathi, M. B. Gawande, R. Zboril and R. S. Varma, Acc. Chem. Res., 2014, 47, 1338 CrossRef PubMed.
- J. D. Mason and S. S. Murphree, Synlett, 2013, 24, 1391 CrossRef CAS PubMed.
- (a) M. Arifuddin, N. Lakshmikant, N. Rajasekar and D. B. Shinde, Indian J. Chem., 2012, 51B, 1168 CAS; (b) A. Amira, H. K'tir, M. Berredjem and N.-E. Aouf, Monatsh. Chem., 2014, 145, 509 CrossRef CAS PubMed.
- T. W. Greene and P. G. M. Wuts, Protecting Group in Organic Synthesis, John Wiley and Sons, New York, 1999 Search PubMed.
- A. Isidro-Llobet, M. Álvarez and F. Albericio, Chem. Rev., 2009, 109, 2455 CrossRef CAS PubMed.
- T. W. Greene and P. G. M. Wuts, Greene's Protective Groups in Organic Synthesis, Wiley & Sons, New York, 4th edn, 2007 Search PubMed.
- C. Supuran, A. Casini and A. Scozzafava, Med. Res. Rev., 2003, 23, 535 CrossRef CAS PubMed.
- A. Scozzafava, T. Owa, A. Mastrolorenzo and C. T. Supuran, Curr. Med. Chem., 2003, 10, 925 CrossRef CAS.
- J. B. McMahon, R. J. Gulakowsky, O. S. Weislow, R. J. Schultz, V. L. Narayanan, D. J. Clanton, R. Pedemonte, F. W. Wassmundt, R. W. Buckheit Jr and W. D. Decker, Antimicrob. Agents Chemother., 1993, 37, 754 CrossRef CAS.
- (a) G. Bartoli, R. Dalpozzo, A. De Nino, L. Maiuolo, M. Nardi, A. Procopio and A. Tagarelli, Green Chem., 2004, 6, 191 RSC; (b) A. Procopio, R. Dalpozzo, A. De Nino, L. Maiuolo, M. Nardi and B. Russo, Adv. Synth. Catal., 2005, 347, 1447 CrossRef CAS; (c) A. Procopio, M. Gaspari, M. Nardi, M. Oliverio, A. Tagarelli and G. Sindona, Tetrahedron Lett., 2007, 48, 8623 CrossRef CAS PubMed; (d) A. Procopio, M. Gaspari, M. Nardi, M. Oliverio and M. O. Rosati, Tetrahedron Lett., 2008, 49, 2289 CrossRef CAS PubMed; (e) A. Procopio, M. Gaspari, M. Nardi, M. Oliverio and R. Romeo, Tetrahedron Lett., 2008, 49, 1961 CrossRef CAS PubMed; (f) A. Procopio, G. Das, M. Nardi, M. Oliverio and L. Pasqua, ChemSusChem, 2008, 1, 916 CrossRef CAS PubMed; (g) A. Procopio, P. Costanzo, M. Curini, M. Nardi, M. Oliverio, R. Paonessa and G. Cravotto, Synthesis, 2011, 1, 0073 CrossRef PubMed; (h) M. Nardi, A. Cozza, L. Maiuolo, M. Oliverio and A. Procopio, Tetrahedron Lett., 2011, 52, 4827 CrossRef CAS PubMed; (i) M. Nardi, A. Cozza, A. De Nino and M. Oliverio, Synthesis, 2012, 44, 800 CrossRef CAS PubMed; (j) A. Procopio, P. Costanzo, M. Curini, M. Nardi, M. Oliverio and G. Sindona, ACS Sustainable Chem. Eng., 2013, 1, 541 CrossRef CAS; (k) M. Nardi, G. Sindona, P. Costanzo, M. Oliverio and A. Procopio, Tetrahedron, 2015, 71, 1132 CrossRef CAS PubMed.
- A. Procopio, M. Oliverio, L. Orio and D. Carnaroglio, Green Chem., 2011, 13, 2806 RSC.
- M. Oliverio, P. Costanzo, R. Paonessa, M. Nardi and A. Procopio, RSC Adv., 2013, 3, 2548 RSC.
- S. Ravichandran and E. Karthikeyan, Int. J. ChemTech Res., 2011, 3, 466–470 Search PubMed.
- C. Alp, Ş. Özsoy1, N. A. Alp, D. Erdem1, M. S. Gültekin, Ö. İ. Küfrevioğlu1, M. Şentürk and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2012, 27(6), 818 CrossRef CAS PubMed.
- R. Almansa, C. fBehloul, D. Guijarro and M. Yus, ARKIVOC, 2007, VII, 41 Search PubMed.
- M. L. Di Gioia, A. Barattucci, P. Bonaccorsi, A. Leggio, L. Minuti, E. Romio, A. Temperini and C. Siciliano, RSC Adv., 2014, 4, 2678 RSC.
- E. C. Roos, P. Bernabe, H. Hiemstra, W. N. Speckamp, B. Kaptein and W. H. J. Boesten, J. Org. Chem., 1995, 60(6), 1733 CrossRef CAS.
- K. J. M. Beresford, N. J. Church and D. W. Young, Org. Biomol. Chem., 2006, 4, 2888 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra16683c |
| This journal is © The Royal Society of Chemistry 2015 |