
The effective adsorption and decomposition of N2O on Al-decorated graphene oxide under electric field
Zhu Lv,
Huiyu Mo,
Chi Chen*,
Xiao Ji,
Kui Xu,
Ling Miao and
Jianjun Jiang
School of Optical and Electronic Information, Huazhong University of Science and Technology, Wuhan, HUBEI 430074, People's Republic of China. E-mail: chenchi_wh89@hotmail.com
First published on 28th January 2015
Abstract
The adsorption property and decomposition process of a N2O molecule on Al-decorated graphene oxide (Al@GO) are investigated using first-principles calculations. The physically adsorbed N2O could be decomposed to N2 molecule and O atom bonded to Al@GO exothermally (2.33 eV per N2O molecule), indicating a stronger interaction of the Al cation and O anion of N2O from that of the reactants. This interaction is enhanced with a positive external electric field, inducing the corresponding higher binding energy and shortened dAl–O. The decomposition barrier of N2O on Al@GO is about 0.50 eV. In particular, due to the elongated dAl–O and shortened dO–N1 in the transition state, the decomposition barrier is also decreased monotonously with increasing electric field. It is remarkable that the N2O decomposition process becomes almost unimpeded and spontaneous under a positive electric field of 0.50 V Å−1. Al-decorated graphene oxide is expected to be a promising new candidate for N2O decomposition with enhanced adsorption and easier decomposition process.
1. Introduction
N2O could cause a series of environmental problems such as the ozone hole1 and the greenhouse effect2 and its investigation about its degradation attracts people's attention.3,4 The decomposition of N2O into N2 and O2 is an eco-friendly way to deal with N2O gas. It is a two-step mechanism that includes N2O → N2 + O followed by N2O + O → N2 + O2, and the first step, which is focused on by most scholars, is more pivotal.5,6 In order to overcome the high energy barrier of N2O decomposition,7–10 plenty of studies have been done to find out appropriate catalysts. Platinum heavy metals11–16 and some transition metals17,18 could effectively decompose N2O with a relatively low barrier. It is interesting that Al was found to have similar catalytic effect with noble metals by A. L. Yakovlev et al.5 Notably, I. S. Chopra et al.19 recently have found that Al could also avoid the shortcomings of other catalysts, such as high costs of platinum heavy metals and transition metals, while maintaining good catalytic effectiveness.Graphene oxide (GO) with large specific surface area and good mechanical properties often serves as the catalyst substrate.20 In addition, metal-decorated GO is widely used in gas adsorption and catalytic decomposition.21–23 In these composites, metal atoms or nano-particles are dispersedly and steadily anchored onto the GO surface through the oxygen functional groups. For example, Al-decorated GO could enhance the adsorption of acid gas and avoid being oxidized at the same time,23 and this property is extremely effective for the degradation of N2O in the atmosphere. On the other hand, an external electric field could be a valid way to adjust the molecule adsorption24,25 and bonding state26–29 of the substrate, which could effectively change the corresponding reaction barrier.10,30
In this work, an eco-friendly light metal, Al, is selected to decorate GO to investigate the process of N2O decomposition. The relaxed structure of the adsorbed state and decomposed state, the binding energy, charge transfer, partial density of state and reaction barrier under different electric fields are carried out to discuss N2O decomposition.
2. Methods and model
Our first-principles calculations are performed using the SIESTA code,31,32 based on density functional theory (DFT).33,34 The exchange–correlation energy is calculated within a uniform generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional.35 We adopt a plane wave cutoff of 150 Ry and a k-point mesh of 4 × 4 × 1 in the Monkhorst–Pack36 sampling scheme. The structure optimization of adsorbed and decomposed states is performed by relaxing the forces on all the atoms until the convergence precision of the forces is less than 0.02 eV Å−1.Al-decorated GO is adopted as the substrate to investigate the adsorption and decomposition processes of N2O. Based on the well-known GO model proposed by A. Lerf and J. Klinowski,37,38 our former research has recently found two relatively stable configurations of Al-decorated GO with adjacent double epoxies or adjacent double hydroxyls (type I and II shown in Fig. 1).23 In this work, a 4 × 4 × 1 hexagonal graphene supercell containing 32 carbon atoms is used as the initial model for GO, as shown in Fig. 1(a). We place GO in the xy plane (9.840 Å × 9.840 Å) and set the translation vector along the z direction as 18 Å to ensure that no interaction occurs between the adjacent cell systems.
 | ||
| Fig. 1 (a) The front views of type I and type II and (b) the planar views of type I and type II. | ||
3. Results and discussion
3.1 N2O adsorption
The adsorption of a N2O molecule on Al@GO is investigated in two configurations according to the orientation of N2O, labeled as the O-end and N-end structures, as shown in Fig. 2. The binding energy Eb of N2O on Al@GO is defined as
| Eb = Etot − (Esubstrate + Egas), |
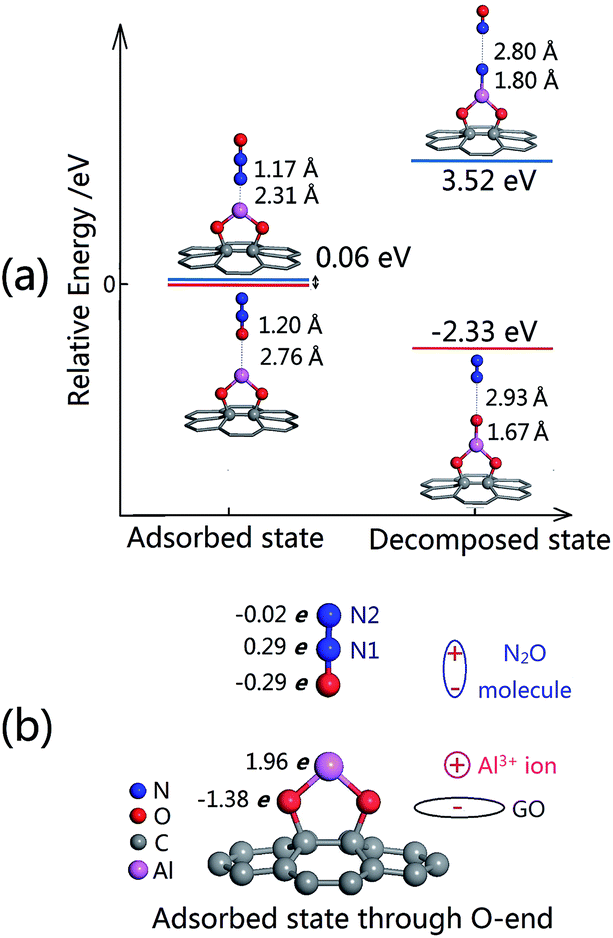 | ||
| Fig. 2 (a) The relative total energies of adsorbed state and decomposed state through N-end and O-end mechanisms and (b) the Bader charge of the adsorbed state through O-end. | ||
The binding energy of N2O to Al@GO through the O-end is −0.25 eV, which is increased significantly compared with that of N2O on pristine graphene (−0.07 eV)10 and similar to that of Al(OH)3(H2O)2 (−0.23 eV).5 The distance between Al and the O atom of the N2O molecule is 2.76 Å, and the lengths of the N1–N2 and O–N1 bonds are almost unchanged, suggesting that the physical adsorption of N2O onto Al@GO is a type of electrostatic interaction. This agrees with the population analysis,39,40 as plotted in Fig. 2(b). An electric dipole moment is formed between the O atom (0.29e) and Al atom (−1.96e), resulting in a relatively strong electrostatic interaction between the N2O molecule and Al@GO.
A N2O molecule could be decomposed into N2 molecule and Oad atom bonded to the Al@GO system. As shown in Fig. 2(a), the distance between the Al atom and O atom is 1.67 Å, showing that a strong chemical bond forms between them. In addition, the distance between the N2 molecule and O atom is 2.93 Å, whereas the bond length of N1–N2 is almost unchanged compared to the previous adsorbed state, leading to a van der Waals adsorption of the N2 molecule onto the O–Al@GO substrate. The abovementioned structural changes reflect old O–N1 bond rupture and new Al–O bond formation in the entire system. In addition, the relative energy of the decomposed state is 2.33 eV lower than that of the adsorbed state, indicating a stronger interaction of the Al–O bond than that of the O–N1 bond. Putting together these analyses, we could look forward to a relatively low decomposition barrier in this reaction.
When a N2O molecule is adsorbed onto the Al@GO through the N-end, we can get a similar adsorbed structure with a slightly higher relative energy (0.06 eV). However, the relative energy of the corresponding decomposed state, in which the Al–N bond forms and N–N bond ruptures, is 3.46 eV higher than that of the adsorbed state. This means that the decomposition reaction of N2O through the N-end on the Al@GO is endothermic and not likely to occur. Therefore, our following works mainly focus on the adsorption and decomposition processes of N2O on the Al@GO through the O-end.
 | ||
| Fig. 3 (a) The binding energies of N2O on Al@GO, (b) dAl–O and dO–N1, and (c) the Bader charges of the Al atom and O atom in adsorbed states under different electric fields. The legend is the same as that of Fig. 2. | ||
The variations of the binding energy and the bond length could result from two dominant factors: the interaction between the inherent electric dipole P with E and the charge redistribution induced by E. The formula W = −P × E means a stronger interaction with increasing E. On the other hand, according to the Bader charge analysis, the charge of the O atom is almost unchanged at the beginning, whereas the charge of the Al atom increases monotonously with increasing E, as shown in Fig. 3(c). A greater electric dipole P induced by the charge transfer could also enhance the interaction between the N2O molecule and Al@GO.
It is noteworthy that when E = 0.50 V Å−1, the charge of the O atom and Al atom changes suddenly. The corresponding dO–N1 (1.24 Å) and dAl–O (2.09 Å) indicate that the old O–N1 bond is weakened significantly and a new Al–O bond is formed. The coexistence of O–N1 and Al–O bonds in the relaxed adsorbed state is very similar to the characteristics of the transition state in decomposition process. Thus the structural transformation from adsorbed state to transition state is favorable with a lower energy barrier under an appropriate electric field.
3.2 N2O decomposition
 | ||
| Fig. 4 (a) Minimum-energy pathway via the N2O → N2 + Oad route, (b) the variations of dAl–O and dO–N1 in the decomposition process, and (c) the Bader charges of the Al atom and O atom in the decomposition process. The legend is the same as that of Fig. 2. | ||
It is important to understand the reaction process and the N2O decomposition mechanism. Here, the initial state, the transition state and the final state are denoted as IS, TS and FS, respectively, in the following discussion. With the reaction proceeding, dAl–O is shortened, whereas dO–N1 is elongated gradually, particularly when it comes to the TS. Compared with the IS, the dAl–O of the TS is shortened from 2.76 Å to 1.92 Å, whereas dO–N1 is lengthened to 0.07 Å, as shown in Fig. 4(b), which means that the structure of the N2O molecule has been affected and the interaction between the Al atom and O atom is strengthened significantly. According to the Bader charge analysis, the charge transfer mainly occurs in the process from the IS to TS, where Al acting as a bridge could transfer the charges from the substrate to the N2O molecule. There is almost no charge redistribution from the TS to FS, as shown in Fig. 4(c), suggesting that the structure of the TS is close to that of the stable FS.
The energy barrier of the N2O decomposition process on the Al@GO structure (0.50 eV) is lower than that of Al(OH)3(H2O)2 (0.87 eV),5 suggesting that the reaction on the Al@GO structure could be performed more easily. The Al atom acts as the catalytic site in both structures, and the chemical state of the Al atom is the dominating factor in the energy barrier to N2O decomposition. In addition, different chemical environments significantly influence the valence and the chemical activity of the Al atom. Therefore, the chemical activity of the Al atom surrounded by three hydroxyl groups in Al(OH)3(H2O)2 is weakened severely because of small effective contact area with the N2O molecule. In comparison, the Al atom in the Al@GO structure only forms a bond with two oxygen atoms of the substrate with relatively larger effective contact area exposed. On the other hand, the reaction heat from the N2O decomposition process on the Al@GO structure (2.33 eV) is larger than that of Al(OH)3(H2O)2 (0.77 eV), which also shows that it is more favorable for N2O to be decomposed on the Al@GO structure.
In order to further investigate the electronic hybridization behavior of N2O decomposition on Al@GO, the partial density of state (PDOS) for three states (IS, TS and FS) are plotted in Fig. 5. As shown in Fig. 5(a), there are two overlapped hybridization orbital peaks between O-2p and N1-2p at about −8.00 eV and 3.00 eV, indicating that the O atom and N1 atom are still strongly covalent bonded. Furthermore, the result that almost no overlapped hybridization orbital peaks between O-2p and N1-2p in Fig. 5(c) proves that the covalent bond between the O atom and N1 atom has been fractured in the FS. Particularly, the PDOS of the TS in Fig. 5(b) shows that the overlapped hybridization orbital peaks between O-2p and Al-3p is weaker than that of the FS, and the similar peaks between O-2p and N1-2p is weaker than that of the IS, suggesting that a new Al–O bond is formed and the old O–N1 bond is gradually fractured.
 | ||
| Fig. 5 PDOS of IS, TS and FS during N2O decomposition process on the Al@GO system. The legend is the same as that of Fig. 2. | ||
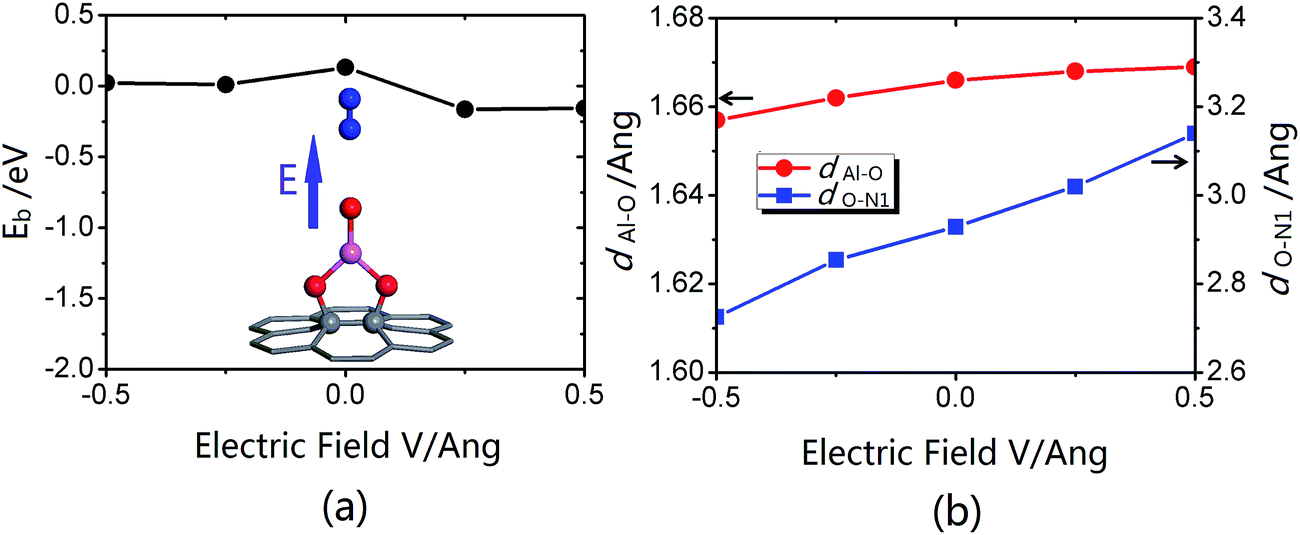 | ||
| Fig. 6 (a) The binding energies, (b) dAl–O and dO–N1 in decomposed states under different electric fields. The legend is the same as that of Fig. 2. | ||
Considering that the analysis of the decomposition barrier could contribute to understanding the role that an electric field plays in the process of N2O decomposition, we calculated the decomposition barrier under each electric field. As shown in Fig. 7, the decomposition barrier is decreased monotonously with E varied from −0.50 V Å−1 to 0.50 V Å−1. It is worth noting that up to E = 0.50 V Å−1, the barrier is almost nonexistent, as the corresponding barrier drops to only 0.02 eV. Furthermore, when E is slightly larger than 0.50 V Å−1, the O–N1 bond is fractured with no barrier and the spontaneous decomposition of N2O on Al@GO could be realized.
 | ||
| Fig. 7 The decomposition barriers and IS, TS, FS under different electric fields. The legend is the same as that of Fig. 2. | ||
To further study the decomposition barrier variation, we investigated the IS, TS and FS under different E, as shown in Fig. 7. As for the TS, dAl–O increases monotonously, whereas dO–N1 decreases monotonously with increasing E. Moreover, the structure of the IS is more similar to that of the TS, contributing to the decrease of the decomposition barrier.
The PDOS of the IS, TS and FS under different electric fields are plotted in Fig. 8. The results show that the PDOSs of every TS and FS almost remains unchanged, whereas those of the corresponding IS exhibit some different characteristics, indicating that an electric field has relatively greater influence on the IS. Furthermore, when E = 0.50 V Å−1, the PDOS of the IS with overlapped Al-3p and O-2p orbital peaks is more analogous to that of the TS, which is consistent with the similarity of their structures. As analyzed above, the electric field could indeed promote the catalytic decomposition of N2O into N2 and Oad on the Al@GO system.
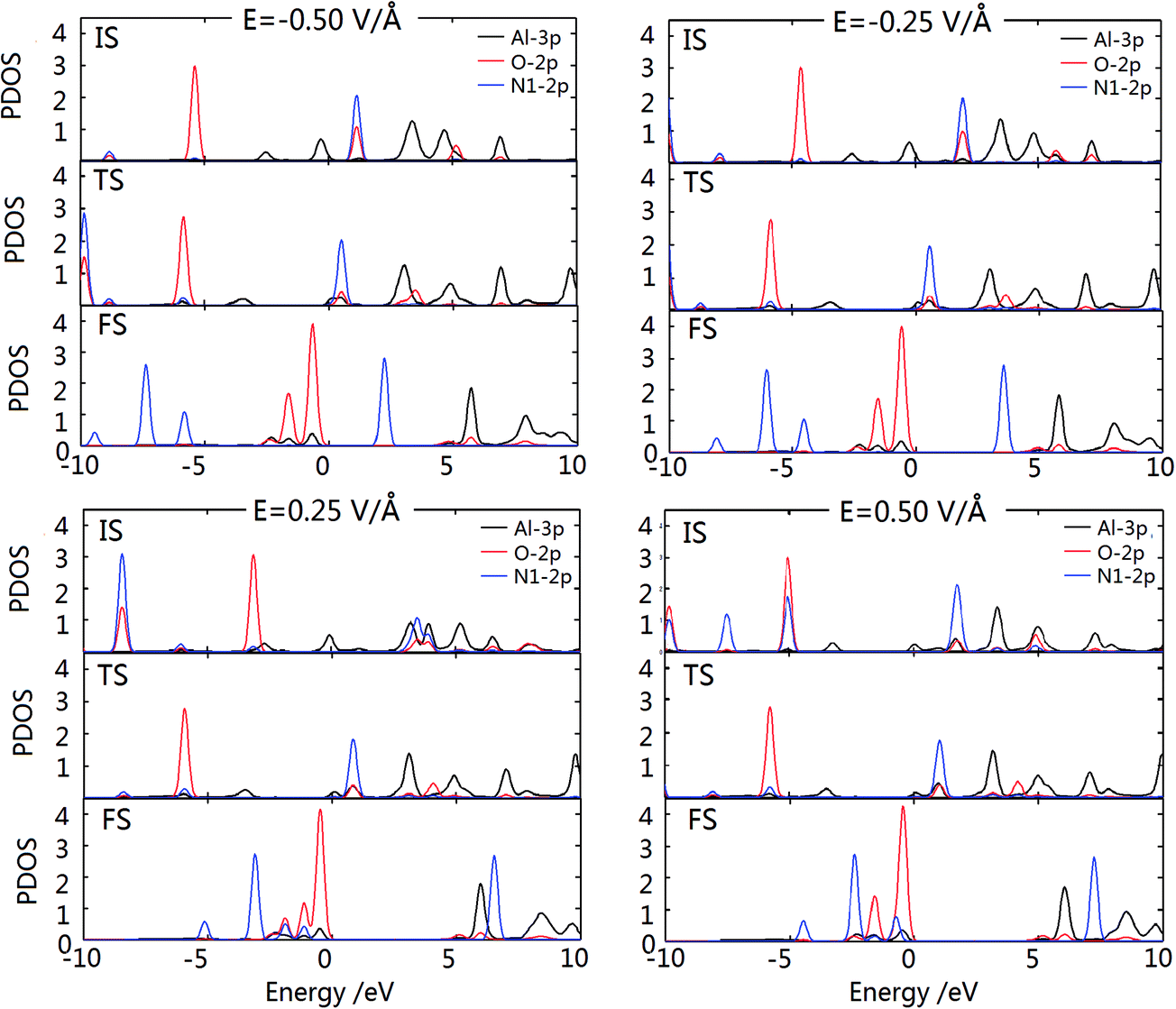 | ||
| Fig. 8 PDOS of IS, TS and FS under different electric fields on the Al@GO system. | ||
4. Conclusions
In summary, the adsorption property and decomposition process of a N2O molecule on Al@GO are investigated by using first-principles calculations. The N2O molecule is adsorbed onto Al@GO through the O-end, and an electric dipole moment is formed between the O atom (0.29e) and Al atom (−1.96e), resulting in a relatively strong electrostatic interaction of the N2O molecule and Al@GO with a binding energy of −0.25 eV. In addition, Al@GO is demonstrated to promote N2O decomposition with considerably lower energy barrier of 0.50 eV. Furthermore, the adsorption and decomposition of N2O on the Al@GO is enhanced with the increasing of a positive external electric field. It should be noted that the N2O decomposition process becomes almost unimpeded and spontaneous under a positive electric field of 0.50 V Å−1 with an energy barrier of 0.02 eV. On the other hand, a van der Waals correction has been taken into consideration, and the results indicate that the influence of van der Waals interactions may be relatively small and the main conclusions keep the same. Thus, our work provides an efficient and economic method for capturing and decomposing N2O gas.Acknowledgements
This research work is supported by the National Natural Science Foundation of China (grant no. 51302097), Wuhan Planning Project of Science and Technology (no. 2013011801010594), the Fundamental Research Funds for the Central Universities, HUST (grant no. CXY13Q003). Computational resources provided by Center of Computational Material Design and Measurement Simulation, Huazhong University of Science and Technology are gratefully acknowledge.References
- A. R. Ravishankara, J. S. Daniel and R. W. Portmann, Science, 2009, 326, 123–125 CrossRef CAS PubMed.
- J. Hansen and M. Sato, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16109–16114 CrossRef CAS PubMed.
- D. J. Wuebbles, Science, 2009, 326, 56 CrossRef CAS PubMed.
- D. J. Beerling, A. Fox, D. S. Stevenson and P. J. Valdes, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9770–9775 CrossRef CAS PubMed.
- A. L. Yakovlev and G. M. Zhidomirov, Catal. Lett., 1999, 63, 91–95 CrossRef CAS.
- E. V. Kondratenko and J. Pérez-Ramírez, Catal. Lett., 2003, 91, 3–4 CrossRef.
- F. Kapteijn, J. Rodriguez-Mirasol and J. A. Moulijn, Appl. Catal., B, 1996, 9, 25–64 CrossRef CAS.
- V. K. Tzitzios and V. Georgakilas, Chemosphere, 2005, 59, 887–891 CrossRef CAS PubMed.
- J. P. Dacquin, C. Dujardin and P. Granger, J. Catal., 2008, 253, 37–49 CrossRef CAS PubMed.
- Y. Lv, G. Zhuang, J. Wang, Y. Jia and Q. Xie, Phys. Chem. Chem. Phys., 2011, 13, 12472–12477 RSC.
- S. Haq and A. Hodgson, Surf. Sci., 2000, 463, 1–10 CrossRef CAS.
- A. Kokalj, I. Kobal, H. Horino, Y. Ohno and T. Matsushima, Surf. Sci., 2002, 506, 196–202 CrossRef CAS.
- R. Burch, S. T. Daniells, J. P. Breen and P. Hu, J. Catal., 2004, 224, 252–260 CrossRef CAS PubMed.
- A. Kokalj and T. Matsushima, J. Chem. Phys., 2005, 122, 034708 CrossRef PubMed.
- F. Rondinelli, N. Russo and M. Toscano, J. Chem. Theory Comput., 2008, 4, 1886–1890 CrossRef CAS.
- X. Wei, X. F. Yang, A. Q. Wang, L. Li, X. Y. Liu, T. Zhang, C. Y. Mou and J. Li, J. Phys. Chem. C, 2012, 116, 6222–6232 CAS.
- A. Stirling, J. Am. Chem. Soc., 2002, 124, 4058–4067 CrossRef CAS PubMed.
- H. Orita and N. Itoh, Surf. Sci., 2004, 550, 166–176 CrossRef CAS PubMed.
- I. S. Chopra, S. Chaudhuri, J. F. Veyan and Y. J. Chabal, Nat. Mater., 2011, 10, 884–889 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- L. Wang, K. Lee, Y. Y. Sun, M. Lucking, Z. Chen, J. J. Zhao and S. B. Zhang, ACS Nano, 2009, 3, 2995–3000 CrossRef CAS PubMed.
- L. Wang, J. Zhao, L. Wang, T. Wang, Y. Sun and S. B. Zhang, Phys. Chem. Chem. Phys., 2011, 13, 21126–21131 RSC.
- C. Chen, K. Xu, X. Ji, L. Miao and J. Jiang, Phys. Chem. Chem. Phys., 2014, 16, 11031–11036 RSC.
- W. Liu, Y. H. Zhao, Y. Li, E. J. Lavernia and Q. Jiang, Phys. Chem. Chem. Phys., 2009, 11, 9233–9240 RSC.
- Z. M. Ao and F. M. Peeters, Appl. Phys. Lett., 2010, 96, 253106 CrossRef PubMed.
- C. He, P. Zhang, Y. F. Zhu and Q. Jiang, J. Phys. Chem. C, 2008, 112, 9045–9049 CAS.
- W. Liu and Q. Jiang, J. Comput. Theor. Nanosci., 2010, 7, 2225–2261 CrossRef CAS PubMed.
- N. Liu, B. Chen, Y. Li, R. Zhang, X. Liang, Y. Li and Z. Lei, J. Phys. Chem. C, 2011, 115, 12883–12890 CAS.
- C. Chen, L. Miao, K. Xu, J. Yao, C. Li and J. Jiang, Phys. Chem. Chem. Phys., 2013, 15, 6431–6436 RSC.
- E. H. Song, J. M. Yan, J. S. Lian and Q. Jiang, J. Phys. Chem. C, 2012, 116, 20342–20348 CAS.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745 CrossRef CAS.
- G. Román-Pérez and J. M. Soler, Phys. Rev. Lett., 2009, 103, 096102 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- B. Hammer, L. B. Hansen and J. K. N. ørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- H. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53–56 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- V. A. Kondratenko and M. Baerns, J. Catal., 2004, 225, 37–44 CrossRef CAS PubMed.
- J. F. Paul, J. P. Ramírez, F. Ample and J. M. Ricart, J. Phys. Chem. B, 2004, 108, 17921–17927 CrossRef CAS.
- J. M. Ricart, F. Ample, A. Clotet, D. Curulla, J. W. Niemantsverdriet, J. F. Paul and J. Pérez-Ramírez, J. Catal., 2005, 232, 179–185 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |