
Lanthanide-supported molybdenum–vanadium oxide clusters: syntheses, structures and catalytic properties†
Fei Fei,
Haiyan An*,
Changgong Meng,
Lin Wang and
Huilong Wang
College of Chemistry, Dalian University of Technology, Dalian 116023, P. R. China. E-mail: anhy@dlut.edu.cn; Tel: +86-411-84657675
First published on 27th January 2015
Abstract
Three new lanthanide-supported molybdovanadates [Ln(H2O)5]2Mo6V2O26·8H2O (Ln = La 1; Ce 2; Nd 3) have been synthesized by the reaction of the [Mo6V2O26]6− anion with Ln(NO3)3 in aqueous solution and characterized by elemental analysis, IR, solid state UV-vis spectroscopy, thermal gravimetric analysis, powder X-ray diffraction and single-crystal X-ray diffraction. Compounds 1–3 are obtained at the same pH value (3.0) and have similar structural units which consist of one molybdovanadate unit supported by two lanthanide cations. Then, these bi-supporting subunits are joined together by strong hydrogen-bonding interactions between polyoxoanions and coordinated water molecules to form a 3D supramolecular framework. To the best of our knowledge, they represent the first examples of inorganic species constructed from molybdenum–vanadium polyoxometalates and lanthanides. The UV-vis diffuse reflectivity spectra of 1–3 show that they can be regarded as a wide gap semiconductor. Photocatalytic studies indicate that compounds 1–3 are not only active photocatalysts for degradation of rhodamine B, but very stable and easily separated from the photocatalytic system for reuse as well. Moreover, the new materials were tested as catalysts in cyanosilylation reactions of aldehydes under solvent-free conditions. The experimental results indicate that all three compounds 1–3 can act as Lewis acid–base catalysts through a heterogeneous manner to prompt cyanosilylation with good efficiency.
Introduction
Polyoxometalates (POMs) as anionic metal oxide clusters, bear many properties that make them attractive for applications in catalysis, medicine, electronics, magnetism and optics.1 Most attention has been given to polyoxotungstates, polyoxomolybdates or polyoxovanadates in this field,2–4 however, it is becoming clear that the molybdenum–vanadium polyoxometalate systems which fuse the merits of molybdenum and vanadium elements, can be specially interesting in catalysis fields. Indeed, some papers have been devoted to the study of homogeneous and heterogeneous catalytic reactions by the molybdenum–vanadium polyoxometalates, such as {PVxMo12−xO40} (x = 0–3).5 Introducing additional metal subunits into molybdenum–vanadium polyoxometalate clusters, will not only build new topologic structures, but also tune the chemical and physical properties of polyoxometalate precursors, thus, always is the subject of intense study. So far, limited success has been achieved in directly introducing metal moieties into molybdenum–vanadium polyoxometalate frameworks,6,7 including 1D chain-like complexes such as [PMo8V6O42][Co(Phen)2][Hpy]·3H3O·H2O and [PMo8V6O42][Co(Phen)2][Him]2·2H3O·3H2O (phen = 1,10-phenanthroline, py = pyridine, Him = imidazole),8 2D layer complexes such as [Co(en)2][Co(bpy)2]2[PMo8V8O44]·4.5H2O and [Cu(en)2(H2O)]{PMo8V6O42[Cu(en)2][Cu0.5(en)]3}·5.5H2O (en = ethylenediamine, bpy = 2,2′-bipyridine),9 and the 3D framework complexes such as {AsMo12O40(VO)[VO(H2O)]}·[Cu(4,4′-bipy)]5·H2O, and Na5Cu(Hmorph)3[V2Mo16O58]·15H2O,10 which have focused on 3d metal cations.Interest in lanthanides with high coordination numbers and flexible coordination geometries has grown significantly in recent years, because lanthanide-based species have emerged as excellent candidates for catalytic studies.11 Lanthanides incorporating into the lacunary polyoxometalates, have been demonstrated as effective Lewis acids for activation of substrates. For example, several lanthanide-containing polyoxometalates [α-Ln(H2O)4P2W17O61]6− and [α-Ln(H2O)PW11O39]4− (Ln = Y3+, La3+, Eu3+, Sm3+, or Yb3+) have been reported to act as Lewis acid catalysts for aldol and imino Diels–Alder reactions.12 To date only two examples of lanthanide-containing molybdenum–vanadium polyoxometalates determined by X-ray single-crystal diffraction are known, i.e. [Ln4(pdc)4(H2O)16][HPMo10V2O40]·H2O (Ln = La, Ce, Pr, Nd, Sm; H2pdc = pyridine-2,6-dicarboxylic acid),13 and [Dy(HL) (L)1.5(H2O)3][PMo11VO40]·8H2O (L = 1,4-bis(pyridinil-4-carboxylato)-l,4-dimethylbenzene).14 Such cases show the difficulty in obtaining the high-quality single crystals containing both constituents. Therefore, it is quite appealing and challenging to directly combining lanthanides with electronically interesting molybdenum–vanadium polyoxometalates to make new materials featuring catalytic functions.
Cyanosilylation reaction provides a convenient route to cyanohydrins, which are key derivatives in the synthesis of fine chemicals and pharmaceuticals.15 In order to obtain cyanohydrin molecules, the use of a catalyst with Lewis acid or base character through which both the carbonyl substrate and the cyanide precursor are activated is necessary. There are very rare examples of cyanosilylation reaction catalyzing by polyoxometalates up to now. An yttrium-containing silicotungstate dimer, [{Y(H2O)2}2(γ-SiW10O36)2]10−, was firstly reported as a Lewis acid–base catalyst by Mizuno's group at 2012, showing remarkable catalytic performance for cyanosilylation of ketones and aldehydes with trimethylsilyl cyanide (TMSCN).16 Then, a series of isostructural lacunary silicotungstate dimers with lanthanide cations, [{Ln(H2O)2(acetone)}2(γ-SiW10O36)2]10− (Ln = Y3+, Nd3+, Eu3+, Gd3+, Tb3+, or Dy3+) have been synthesized by them and effectively promoted cyanosilylation of carbonyl compounds with trimethylsilyl cyanide as homogeneous catalysts.17 Recently, Niu and coworkers have successfully isolated one [H2W11O38]8−-based metal–organic framework {[Cu2(bpy)(H2O)5.5]2[H2W11O38]·3H2O·0.5CH3CN},18 which can be as a Lewis acid catalyst through a heterogeneous manner to drive cyanosilylation with good efficiency. The development of efficient heterogeneous catalysts for cyanosilylation of carbonyl compounds with trimethylsilyl cyanide still is a significant work in current research. It is thus of considerable interest to investigate whether lanthanide-containing molybdenum–vanadium polyoxometalates can effectively catalyze the cyanosilylation reaction as heterogeneous catalysts.
Herein, we study the interactions between the molybdenum–vanadium oxide cluster [Mo6V2O26]6− and different lanthanide cations. Three new compounds were obtained, namely [Ln(H2O)5]2Mo6V2O26·8H2O (Ln = La 1; Ce 2; Nd 3). Compounds 1–3 are constructed from [Mo6V2O26]6− anions supported by two lanthanide cations, representing the first examples of inorganic species based on molybdenum–vanadium polyoxometalates and lanthanides. The [Mo6V2O26]6− cluster consists of six [MoO6] and two [VO6] octahedra connected by shared edges, isostructural with the octamolybdate anion β-[Mo8O26]6−. Noteworthy, all three compounds as heterogeneous Lewis acid–base catalysts are very efficient in the cyanosilylation of carbonyl compounds and allow the reaction to be carried out under solvent-free conditions using only a low loading of the catalyst, which can be recovered and reused without displaying any significant loss of activity. Furthermore, compounds 1–3 show good activity in photocatalyzing degradation of organic dye RhB.
Experimental section
Materials and measurements
La(NO3)3·6H2O, Ce(NO3)3·6H2O and Nd(NO3)3·6H2O were commercially purchased. The precursor K5NaMo6V2O26·4H2O was prepared as described by Nenner and characterized by IR spectroscopy.19 All chemicals were commercially purchased and used as received without further purification. A Perkin-Elmer CNHS analyzer 2400 was employed for elemental analysis. IR spectra were recorded in the range 400–4000 cm−1 on an Alpha Centaur FT/IR Spectrophotometer using KBr pellets. TG analyses were performed on a Perkin-Elmer TGA7 instrument in flowing N2 with a heating rate of 10 °C min−1. PXRD patterns of the samples were recorded on a Rigaku Dmax 2000 X-ray diffractometer with graphite monochromatized Cu-Kα radiation (λ = 0.154 nm) and 2θ varying from 5 to 50°. Diffuse reflectivity spectra were collected on a finely ground sample with a Cary 500 spectrophotometer equipped with a 110 mm diameter integrating sphere, which were measured from 200 to 800 nm. UV-vis spectra were acquired on an Analytic Jena SPECORD S600 spectrophotometer.Synthesis of [La(H2O)5]2Mo6V2O26·8H2O (1)
In a typical synthesis procedure for 1, K5NaMo6V2O26·4H2O(138.4 mg, 0.1 mmol) and La(NO3)3·6H2O (86.6 mg, 0.2 mmol) were dissolved with stirring in 20 mL deionized water. The pH value of the mixture is about 3.50. After the pH value was adjusted to 3.00 with 1 M HNO3, the solution was heated at 80 °C for 1 h. When the solution was cooled to room temperature, a small amount of pale yellow precipitate was filtered off, and then the filtrate was sealed with pierced parafilm for facilitating slow evaporation at room temperature. The filtrate was kept for one week at ambient conditions, and yellow block crystals of 1 were obtained in about 40% yield (based on V). Anal. calcd (%) for [La(H2O)5]2Mo6V2O26·8H2O: Mo, 33.96; La, 16.39; V, 6.01; O, 41.52; H, 2.12. Found (%): Mo, 33.59; La, 17.96; V, 6.28; O, 40.16; H, 2.01. FTIR data (cm−1): 3377(s), 1626(m), 947(s), 918(s), 890(s), 796(m), 647(s), 563(m), 533(m), 431(w).Synthesis of [Ce(H2O)5]2Mo6V2O26·8H2O (2)
Compound 2 was synthesized by the same procedure as 1 except for replacement of La(NO3)3·6H2O with Ce(NO3)3·6H2O (86.8 mg, 0.2 mmol) and pH = 2.96. The filtrate was kept for one week at ambient conditions, and then yellow block crystals of 2 were isolated in about 30% yield (based on V). Anal. calcd (%) for [Ce(H2O)5]2Mo6V2O26·8H2O: Mo, 33.92; Ce, 16.49; V, 6.01; O, 41.46; H, 2.12. Found (%): Mo, 34.01; Ce, 16.87; V, 5.32; O, 41.81; H, 1.99. FTIR data (cm−1): 3375(s), 1627(m), 948(s), 919(s), 887(s), 797(m), 649(s), 559(m), 527(m), 429(w).Synthesis of [Nd(H2O)5]2Mo6V2O26·8H2O (3)
Compound 3 was synthesized by the same procedure as 1 except for replacement of La(NO3)3·6H2O with Nd(NO3)3·6H2O (87.6 mg, 0.2 mmol) and pH = 3.02. The filtrate was kept for one week at ambient conditions, and then yellow block crystals of 3 were isolated in about 30% yield (based on V). Anal. calcd (%) for [Nd(H2O)5]2Mo6V2O26·8H2O: Mo, 33.76; Nd, 16.88; V, 5.98; O, 41.27; H, 2.11. Found (%): Mo, 33.12; Nd, 17.37; V, 6.26; O, 41.08; H, 2.17. FTIR data (cm−1): 3369(s), 1622(m), 943(s), 918(s), 882(s), 797(m), 634(s), 563(m), 531(m), 428(w).X-ray crystallography
The crystallographic data of three compounds were collected on the Bruker Smart CCD diffractometer with Mo Kα radiation (λ = 0.71073 Å) by ω and θ scan modes. Empirical absorption correction was applied. The structures of 1–3 were solved by the direct method and refined by the Full-matrix least squares on F2 using the SHELXTL-97 software.20 All of the non-hydrogen atoms were refined anisotropically in 1–3. The hydrogen atoms attached to water molecules were not located in 1–3. The detailed crystal data and structure refinements for 1–3 are given in Table 1. Selected bond lengths and angles of 1 and 3 are respectively listed in Table 2. Crystallographic data for the structures reported in this paper have been deposited. CSD reference number: 428893 for 1, 428894 for 2 and 428895 for 3.| Complex | 1 | 2 | 3 |
|---|---|---|---|
| a R1 = ∑‖F0| − |FC‖/∑|F0|.b wR2 = ∑[w(F02 − FC2)2]/∑[w(F02)2]1/2. | |||
| Formula | H36La2Mo6O44V2 | H36Ce2Mo6O44V2 | H36Nd2Mo6O44V2 |
| Formula weight | 1695.63 | 1698.05 | 1706.29 |
| T (K) | 293(2) | 296(2) | 296(2) |
| Crystal system | Triclinic | Triclinic | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
| a (Å) | 10.0427(4) | 10.0173(3) | 10.038(5) |
| b (Å) | 10.1151(4) | 10.0913(3) | 10.049(6) |
| c (Å) | 19.7105(8) | 19.6748(7) | 10.118(6) |
| α (°) | 77.558(2) | 77.372(2) | 106.967(8) |
| β (°) | 82.638(2) | 82.719(2) | 101.480(9) |
| γ (°) | 78.650(2) | 78.668(2) | 91.502(8) |
| U (Å3) | 1909.29(13) | 1895.62(10) | 952.8(9) |
| Z | 2 | 2 | 1 |
| μ (mm−1) | 4.677 | 4.859 | 5.169 |
| Reflections collected | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 896 896 |
9084 | 4586 |
| Independent reflections | 6693 | 6581 | 3293 |
| R (int) | 0.0195 | 0.0189 | 0.0412 |
| GOF on F2 | 1.044 | 1.027 | 1.028 |
| R1a [I > 2σ(I)] | 0.0287 | 0.0373 | 0.0568 |
| wR2b [I > 2σ(I)] | 0.0687 | 0.0937 | 0.1454 |
| R1 (all data) | 0.0362 | 0.0549 | 0.0917 |
| wR2 (all data) | 0.0745 | 0.1087 | 0.1661 |
| a Symmetry transformations used to generate equivalent atoms: #1 −x, −y + 1, −z. | |||
|---|---|---|---|
| Compound 1 | |||
| Mo(2)–O(2) | 1.689(4) | Mo(5)–O(20) | 1.719(4) |
| Mo(5)–O(26) | 1.707(4) | Mo(2)–O(14) | 1.748(4) |
| Mo(4)–O(13) | 1.899(4) | Mo(3)–O(4) | 1.963(4) |
| Mo(2)–O(11) | 2.206(4) | Mo(4)–O(4) | 2.237(4) |
| Mo(4)–O(16) | 2.281(4) | V(2)–O(7) | 1.625(4) |
| Mo(2)–O(1) | 2.343(4) | V(2)–O(1) | 2.227(4) |
| La(1)–O(17) | 2.530(4) | La(2)–O(9) | 2.535(4) |
| La(1)–O(6) | 2.623(4) | La(2)–O(7) | 2.656(4) |
| La(1)–O(1W) | 2.518(4) | La(2)–O(7W) | 2.496(4) |
| La(1)–O(4W) | 2.580(4) | La(2)–O(8W) | 2.593(4) |
| O(16)–V(2)–O(1) | 76.63(13) | O(6)–V(1)–O(18) | 106.10(19) |
| O(19)–V(1)–O(4) | 151.86(15) | O(6)–V(1)–O(16) | 170.29(17) |
| Compound 3a | |||
| Mo(2)–O(7) | 1.680(10) | Mo(3)–O(3) | 1.713(9) |
| Mo(1)–O(6) | 1.695(10) | Mo(2)–O(1) | 1.723(9) |
| Mo(2)–O(13) | 1.965(9) | Mo(3)–O(2) | 1.887(9) |
| Mo(3)–O(5) | 1.969(10) | Mo(1)–O(4) | 1.933(10) |
| Mo(3)–O(11) | 2.280(10) | V(1)–O(10)#1 | 1.634(10) |
| Mo(1)–O(11) | 2.336(9) | V(1)–O(11) | 2.211(9) |
| Nd(1)–O(3) | 2.534(9) | Nd(1)–O(1W) | 2.510(12) |
| Nd(1)–O(10) | 2.632(10) | Nd(1)–O(4W) | 2.586(12) |
| O(11)#1–V(1)–O(11) | 76.8(4) | O(10)#1–V(1)–O(9) | 105.8(5) |
| O(5)#1–V(1)–O(13)#1 | 152.2(4) | O(10)#1–V(1)–O(11) | 169.9(4) |
General procedure for cyanosilylation reactions
The detailed reaction conditions are shown in the captions of Table 3. A typical procedure for the cyanosilylation reaction of aldehydes is as follows: 1 mol% catalyst (1–3) was added to a mixture of aldehyde (0.5 mmol) and (CH3)3SiCN (1.5 mmol), in the absence of solvent. The reaction mixture was stirred at room temperature under a N2 atmosphere. The progress of the reaction was monitored by GC analysis. After the reaction was completed, the catalyst was removed by filtration and centrifugation of the reaction mixture. All products (cyanohydrin trimethylsilyl ethers) were confirmed by a comparison of their GC retention times, GC-MS spectra, and/or 1H spectra with those of authentic data. GC analysis was performed using HP6890/5973MS with a cross-linked (95%)-dimethyl-(5%)-diphenylpolysiloxane column of 30 m length.
|
|||
|---|---|---|---|
| Compound | Aldehyde | Time (h) | Yieldb (%) |
| a Reaction conditions: catalyst 1 mol%, aldehyde 0.5 mmol, TMSCN 1.5 mmol, without solvent, room temperature (25 °C) under N2.b Yields were determined by GC analysis using naphthalene as an internal standard. | |||
| 1 |  |
9 | 93.5 |
| 2 | 9 | 90.3 | |
| 3 | 7 | 96.2 | |
|
|||
| 3 |  |
9 | 78.6 |
| 3 |  |
7 | 92.8 |
| 3 |  |
9 | 90.2 |
| 3 |  |
9 | 70.5 |
| 3 |  |
6 | 96.6 |
| Blank |  |
7 | 16.8 |
| K5NaMo6V2O26·4H2O |  |
9 | 58.4 |
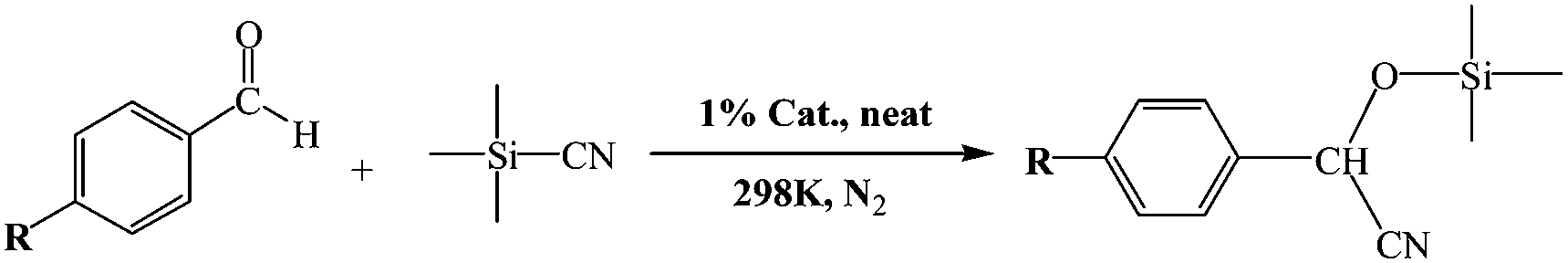
Results and discussion
Synthesis
The reaction of the β-[Mo6V2O26]6− polyoxometalate and three different trivalent lanthanide cations (Ln = La3+, Ce3+, Nd3+) has been investigated. Compounds 1–3 were synthesized under the same reaction conditions (i.e., the same Ln/[Mo6V2O26]6− ratio (2:1), same temperature, same concentration of starting polyoxometalate, and same pH) by the conventional solution method which possesses reasonably good solubility and are environmentally friendly. Many parallel experiments indicate that stoichiometry, pH value and reaction temperature can affect the quality of crystals. The products are obviously affected by the stoichiometry of Ln3+/[Mo6V2O26]6−. Compounds 1–3 can be well isolated by the 2:1 Ln3+/[Mo6V2O26]6− ratio. When the Ln3+/[Mo6V2O26]6− ratio increased from 2:1 to 3:1, 4:1 or decreased to 1:1, the crystal quality of compounds 1–3 was poor, and the yields were reduced. The optimal pH range of the reaction is from 2.7 to 3.2. For a low pH (pH < 2.5) no reaction occurs; for a high pH (pH > 3.5) the hydrolysis of the lanthanide cations makes a mass of precipitates rather than the crystals. The optimal reaction temperature of the reaction is about 80 °C. At higher temperature (such as 100 °C), crystals with poor quality can be obtained; at lower temperature (such as 25 °C), no crystals were observed. Furthermore, when Eu3+, Gd3+ or Tb3+ ions were used instead of La3+, Ce3+ or Nd3+ ions in compounds 1–3 under the similar conditions, only yellow precipitates were got. This phenomenon is attributed to the lanthanide contraction effect, and has been observed in other lanthanide–POM-based compounds.21
Crystal structures of 1–3
Single-crystal X-ray crystallographic analyses reveal that all compounds 1–3 are composed of [Mo6V2O26]6− polyoxoanion to form a bi-supporting structure. The polyoxoanion [Mo6V2O26]6− was first prepared and structurally determined as its pentapotassium sodium salt by Nenner in 1985.19 The structure of [Mo6V2O26]6− anion consists of six [MoO6] and two [VO6] octahedra connected by shared edges, with two [Mo3VO13]3− subunits stacking together, which is isostructural with the octamolybdate anion β-[Mo8O26]4− (shown in Fig. S1†).Isostructural compounds 1 and 2 crystallize in triclinic space group P. Herein compound 1 is described as an example in detail. The asymmetric unit in the structure of 1 consists of one crystallographically independent [Mo6V2O26]6− polyoxoanion, two La3+ units and eight free water molecules (Fig. 1a). In the polyoxoanion, each [MoO6] octahedron has two terminal oxygen atoms and each [VO6] octahedron has one terminal oxygen atom. According to the manner of oxygen coordination, five kinds of oxygen atoms exist in the β-[Mo6V2O26]6− cluster, that is six terminal oxygen Ot, eight terminal oxygen Ot′ linked to La3+, six double-bridging oxygen Ob, four central oxygen Oc (μ3-O atom of a V and two Mo atoms), two central oxygen Od (μ5-O atom of two V and three Mo atoms). Thus the Mo–O bond lengths fall into five classes: Mo–Ot 1.689(4)–1.707(4) Å, Mo–Ot′ 1.719(4)–1.748(4) Å, Mo–Ob 1.899(4)–2.206(4) Å, Mo–Oc 1.963(4)–2.237(4) Å, and Mo–Od 2.281(4)–2.343(4) Å in 1. The two independent vanadium centers V(1) and V(2) possess distorted octahedral environments. The V–O distances vary from 1.625(4) to 2.227(4) Å. The bond angles of O–V–Ocis range from 76.63 to 106.10°, and O–V–Otrans range from 151.86 to 170.29°. All bond lengths and bond angles are within the normal ranges.19
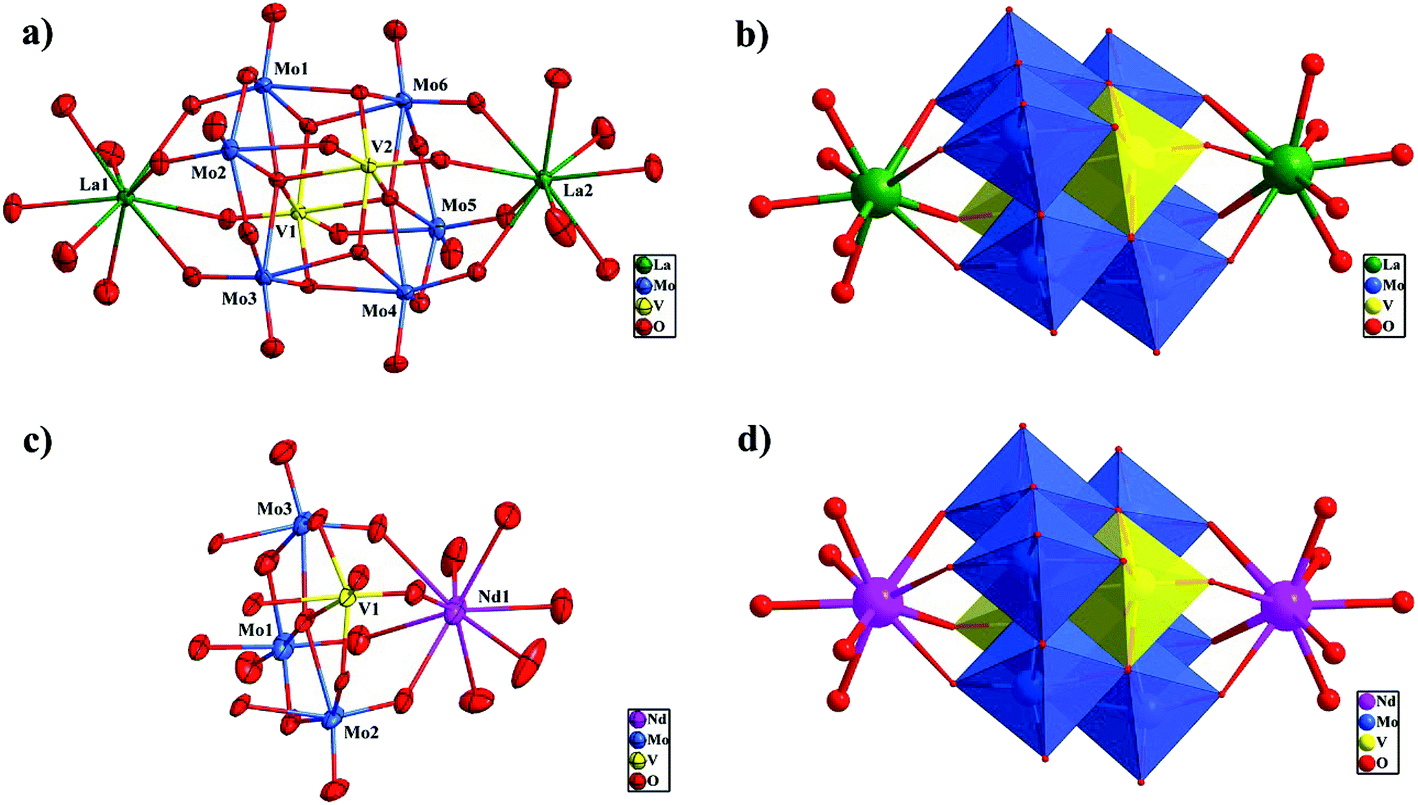 | ||
| Fig. 1 (a) ORTEP drawing of 1 with thermal ellipsoids at 50% probability. (b) Polyhedral and ball-stick representation of one β-[Mo6V2O26]6− polyoxoanion supported by two {La(H2O)6} units in 1. (c) ORTEP drawing of 3 with thermal ellipsoids at 50% probability. (d) Polyhedral and ball-stick representation of one β-[Mo6V2O26]6− polyoxoanion supported by two {Nd(H2O)6} units in 3 (color code: V, yellow; Mo, blue; La, green; Nd, pink; O, red). | ||
Different from compound 1, the asymmetric unit in the structure of 3 consists of half one crystallographically independent [Mo6V2O26]6− polyoxoanion, one Nd3+ unit and four free water molecules (Fig. 1c). Also, five kinds of oxygen atoms exist in the cluster according to the manner of oxygen coordination. Then, the Mo–O bond lengths fall into five classes: Mo–Ot 1.680(1)–1.695(1) Å, Mo–Ot′ 1.713(9)–1.723(9) Å, Mo–Ob 1.887(9)–1.933(9) Å, Mo–Oc 1.965(9)–1.969(1) Å, and Mo–Od 2.280(1)–2.336(9) Å in 3. The V–O distances are 1.634(1)–2.211(9) Å, and the O–V–O angles are in the range of 76.8(4)–169.9(4)°. All bond lengths and bond angles are similar to compound 1.
There are two crystallization-independent La(III) cations display a similar coordination sphere in 1 (Fig. S2†). La(1) cation, coordinating to [Mo6V2O26]6− polyoxoanion, has a monocapped square antiprismatic coordination geometry, defined by four terminal oxygen atoms from one [Mo6V2O26]6− unit [La–O 2.530(4)–2.623(4) Å] and five water molecules [La–OH2 2.518(4)–2.580(4) Å]. The nine-coordinate La(2) also links to four terminal oxygen atoms from [Mo6V2O26]6− molecule [La–O 2.535(4)–2.656(4) Å] and five water molecules [La–OH2 2.496(4)–2.593(4) Å] to complete a monocapped square antiprismatic coordination geometry. The average La–O bond length is 2.562 Å, which is comparable to the sum of La–O ionic radii for nine-coordinate La3+ and two-coordinate O2− ions (2.56 Å).22 In 3, one crystallographically unique Nd3+ ion bonded to the polyoxoanion as supporter, is coordinated by four oxygen atoms from the [Mo6V2O26]6− POM [Nd–O 2.534(9)–2.632(1) Å] and five water molecules [Nd–OH2 2.510(1)–2.586(1) Å] to form a monocapped square antiprismatic coordination geometry (Fig. S3†). The average Nd–O bond length is 2.566 Å, which is closed to the Nd–O bond distances in K0.5Nd0.5[Nd2(SiW11O39) (H2O)11]21a and [NdK(H2O)12][Nd(H2O)6]2[(H2O)4NdBW11O39H]2.23
Interestingly, in compounds 1 and 3 each [Mo6V2O26]6− polyoxoanion is linked by two [Ln(H2O)5]3+ cations to construct a similar bi-supporting structure (Fig. 1b and d). Then, theses bi-supporting subunits are firstly joined up together by the strong hydrogen-bonding interactions (O26⋯O4W 2.802 Å and O2⋯O6W 2.851 Å in 1) between terminal oxygen atoms of polyoxoanions and coordinated water molecules to form a 1D supramolecular chain (shown in Fig. 2a). Then adjacent chains are linked together by other hydrogen-bonding interactions (O12⋯O2W 2.821 Å, O24⋯O3W 2.765 Å, O25⋯O10W 2.857 Å in 1) to generate an interesting 2D supramolecular wavelike network (see Fig. 2b). Furthermore, 2D supramolecular layers are connected to yield a 3D supramolecular channel framework via the strong hydrogen-bonding interactions (O18⋯O1W 2.761 Å, O26⋯O4W 2.914 Å and O11⋯O9W 2.829 Å in 1) (Fig. 2c). Free water molecules are filled in the cavities of 3D supramolecular structures and participate in the extensive hydrogen-bonding interactions with the polyoxoanion framework. A better insight into the nature of such 3D supramolecular framework can be achieved by reducing the structure to simple node and linker net. According to the simplification principle, each bi-supporting cluster [La(H2O)5]2Mo6V2O26 is linked with eight such units by strong hydrogen bonds. Therefore, the resulting 3D supramolelar frame is a rare uninodal 8-connected net, as shown in Fig. S4.†
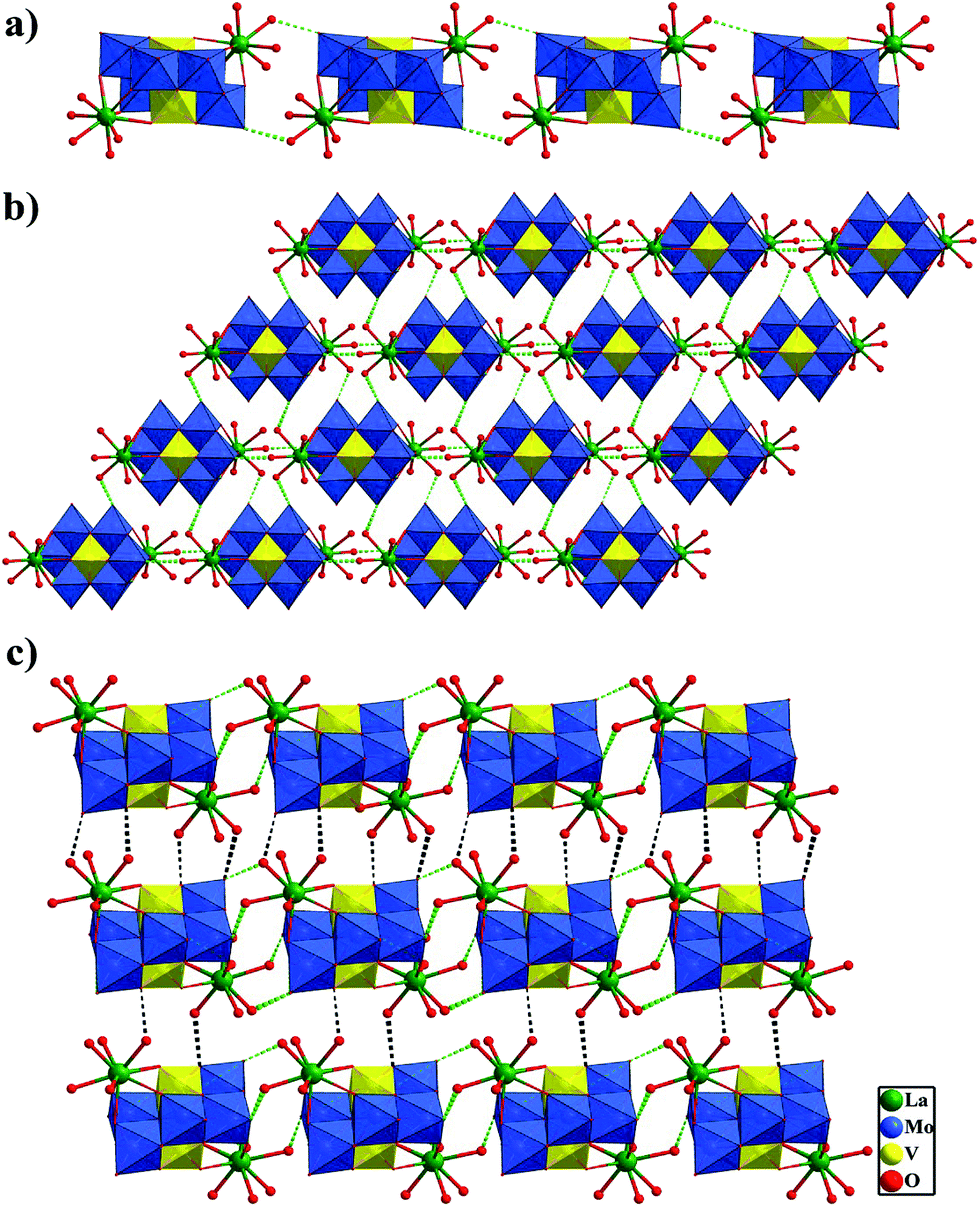 | ||
| Fig. 2 (a) A view of the 1D supramolecular chain in 1, showing the hydrogen bonds between the terminal oxygen atoms of polyoxoanions and coordinating water molecules with green color. (b) 2D supramolecular layer in 1, showing the hydrogen bonds between polyoxoanions and coordinated water molecules with green color. (c) 3D supramolecular framework in 1, displaying the interbedded hydrogen bonds between polyoxoanions and coordinated water molecules with black color (color code: V, yellow; Mo, blue; La, green; O, red). Free water molecules are omitted for clarity. | ||
Bond valence sum calculations24 show that all Mo atoms are in the +6 oxidation state, V atoms are in the +5 oxidation state, and lanthanide sites are in the +3 oxidation state. These results are consistent with the charge balance considerations.
FT-IR spectra
The IR spectra of compounds 1–3 display similar characteristic patterns of the [Mo6V2O26]6− structure (see Fig. S5a–S5c†). The characteristic bands of ν(M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ot) vibrations are observed at 947, 918 cm−1 for 1, 948, 919 cm−1 for 2 and 943, 918 cm−1 for 3; the characteristic bands of ν(M–Ob–M) and ν(M–Oe–M) are at 890, 796, 647 cm−1 for 1, 887, 797, 649 cm−1 for 2 and 882, 797, 634 cm−1 for 3 (M = Mo or V; Oc – corner shared oxygen; and Oe – edge shared oxygen).10b,25 Below 600 cm−1, the spectrum is rather difficult to analyze, the bending of the M–Ot and M–Ob bonds can be mixed with the M–Oe stretching vibrations. These characteristic bands are nearly identical to those of the reported octamolybdate compounds except for slight shifts of some peaks. A broad band around 3377 cm−1 of 1, 3375 cm−1 of 2 and 3369 cm−1 of 3 is attributed to the absorptions of water molecules.
Ot) vibrations are observed at 947, 918 cm−1 for 1, 948, 919 cm−1 for 2 and 943, 918 cm−1 for 3; the characteristic bands of ν(M–Ob–M) and ν(M–Oe–M) are at 890, 796, 647 cm−1 for 1, 887, 797, 649 cm−1 for 2 and 882, 797, 634 cm−1 for 3 (M = Mo or V; Oc – corner shared oxygen; and Oe – edge shared oxygen).10b,25 Below 600 cm−1, the spectrum is rather difficult to analyze, the bending of the M–Ot and M–Ob bonds can be mixed with the M–Oe stretching vibrations. These characteristic bands are nearly identical to those of the reported octamolybdate compounds except for slight shifts of some peaks. A broad band around 3377 cm−1 of 1, 3375 cm−1 of 2 and 3369 cm−1 of 3 is attributed to the absorptions of water molecules.
TG analysis
Thermal gravimetric (TG) curve of 1 is shown in Fig. S6a.† The TG curve possesses a two-step continuous weight loss process in the range of 40–600 °C and gives a total loss of 18.5%, which arises from the loss of all lattice water molecules and coordinated water molecules and agrees with the calculated value of 19.1%. The TG curves of 2 and 3 exhibit similar weight loss stages to those of 1 (see Fig. S6b and S6c†), and give a total weight loss of 18.4% and 18.9% in the range of 40–600 °C, which agree with the calculated value of 19.1% and 19.0%.Powder X-ray diffraction characterization
The PXRD patterns for 1–3 are presented in Fig. S7.† The diffraction peaks of both calculated and experimental patterns match well, indicating the phase purities of these compounds. These conclusions are in agreement with the results of the single crystal X-ray analysis.Optical band gap
To investigate the conductivity potentials of compounds 1–3, the diffuse reflectivity for solid samples of 1–3 were performed to obtain the band gap (Eg). The absorption (α/S) data were calculated from the reflectivity using the Kubelka–Munk function: α/S = (1 − R)2/2R where R is the reflectivity at a given wavelength, α is the absorption coefficient, and S is the scattering coefficient. The band gap was determined as the intersection point between the energy axis at α/S = 0 and the line extrapolated from the linear portion of the adsorption edge in a plot of α/S against energy E. The α/S versus E plots for compounds 1–3 are shown in Fig. 3. For compounds 1–3, the well-defined optical absorption associated with band gaps (Eg) can be assessed at 2.87, 2.91 and 2.94 eV, respectively. The reflectance spectra imply that compounds 1–3 are potential semiconductors with wide band gaps. In addition, the optical band gap of the precursor K5NaMo6V2O26·4H2O has been measured (Fig. S8†). The Eg value of the precursor is 3.17 eV, which is higher than those of 1–3. Therefore, the optical band gaps of 1–3 are relevant to the POM β-[Mo6V2O26]6− and lanthanide cations, revealing that the optical band gaps of POM-based compounds could be tuned effectively via metal cations. | ||
| Fig. 3 UV-vis diffuse reflectivity spectra of K–M functions vs. energy (eV) of compounds 1–3. | ||
Photocatalytic activity
The evaluation of photocatalytic activities of three samples 1–3 for the photodegradation of rhodamine B (RhB) was performed at ambient temperature (25 °C). The procedure was as follows: a sample (10.0 mg) was mixed with 50 mL of 2 × 10−5 mol L−1 RhB solution, followed by magnetically stirring in the dark for about 30 min. The solution was then exposed to UV irradiation from a 300 W Hg lamp in the photocatalytic reaction instrument, kept stirring during irradiations. A sample was taken out every 1 h for analysis. The evolution of the spectral changes taking place during the photodegradation of RhB over 1–3 is shown in Fig. 4. It can be clearly seen that the absorbance peaks of RhB are decreased obviously for 1–3 with increasing reaction time. The calculations reveal that the conversions of RhB are 77.5% for 1, 75.6% for 2 and 74.3% for 3 after 9 h. In comparison, the absorption peaks of RhB without any catalyst show no obvious change (Fig. S9†). The results indicate that compounds 1–3 show good photocatalytic activities for the degradation of RhB. Under the same condition, the precursor K5NaMo6V2O26·4H2O of the same weight as homogeneous catalyst was used to photocatalytically decompose the RhB, resulting in drop of the RhB concentrations by 53.4% after 9 h (Fig. S10†), so the introduction of lanthanide cations can play the role of improving the photocatalytic activity. Niu's group has synthesized many new compounds based on lanthanides and lacunary Keggin POMs, and studied their photocatalytic activities on degrading the RhB under UV irradiation.26 Their experimental results indicate that the lanthanide cations may play an important role in inhibiting the photodegradation of RhB. Compounds 1–3 represent the first examples that lanthanide-based POMs as photocatalysts enhance the degradation of organic dye up to now, although their photocatalytic conversions are moderate under UV irradiation. According to the literatures,27 POM-based compounds have a similar electronic attribution to that of semiconductors such as TiO2. Both POM and TiO2 have d0 transition metal atoms and oxygen atoms. UV irradiation of 1–3 resulted in the formation of an O → M charge-transfer excited state (M = Mo, V), producing considerable holes and electrons, which can be considered as a parallel process to the promotion of an electron from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO).28 The photoinduced holes can oxidize RhB molecules in the solution. Thus, compounds 1–3 became a powerful oxidant by UV irradiation. Furthermore, compounds 1–3 as heterogeneous catalysts can be easily isolated from the reaction suspension by simple filtration (Fig. S11†). To demonstrate their recyclability, successive reactions were performed, showing that the activity of the recovered catalyst does not decrease after four cycles at least (Fig. S12†). | ||
| Fig. 4 (a) Absorption spectra of the RhB solution during the decomposition reaction under UV irradiation in the presence of compound 1. (b) Absorption spectra of the RhB solution during the decomposition reaction with catalyst 2. (c) Absorption spectra of the RhB solution during the decomposition reaction with catalyst 3. (d) Photocatalytic decomposition rate of the RhB solution under UV irradiation with the use of K5NaMo6V2O26·4H2O, compounds 1–3 and only RhB. | ||
Cyanosilylation study
The catalytic properties of three compounds 1–3 as heterogeneous catalysts in the cyanosilylation reactions of aldehydes have been investigated under solvent-free condition at room temperature. Firstly, the cyanosilylation of benzaldehyde with trimethylsilyl cyanide (TMSCN) acts as a model reaction to investigate the catalytic nature of compounds 1–3. The reaction was carried out with 1 mol% of catalyst under the conditions described in Table 3. All three compounds effectively catalyzed the cyanosilylation to afford the corresponding cyanohydrin trimethylsilyl ether, and a small difference in activity was found among them (see Fig. S13†). The yield of Nd-based compound 3 is 96.2%, followed by La-based compound 1 (93.5%) and Ce-based compound 2 (90.3%). In a comparison of the activity of our catalysts with that of some previously reported POM-based compounds ([Cu2(bpy)(H2O)5.5]2[H2W11O38],18 and TBA8H2[(SiYW10O36)2]),16 it was found to be higher than the first one (benzaldehyde, 24 h, and yield = 98.1%), which uses solvent conditions, and less than the second one, which is a homogeneous catalyst and uses solvent conditions (benzaldehyde, 15 min, and yield = 94%).Subsequently, the cyanosilylation reaction of various kinds of aromatic and aliphatic aldehyde substrates in the presence of Nd-based catalyst 3 was further studied (Fig. S14†). The effect of the nature of the aldehyde was evidenced that the activity of aliphatic aldehyde such as heptaldehyde is higher than the aromatic aldehydes like 2-hydroxybenzaldehyde, 4-fluorobenzaldehyde, or 4-methylbenzaldehyde (Table 3). When sterically more hindered 1-napthaldehyde is used as the substrate, the yield is even poor suggesting that the aldehyde here is not accommodated as per the transition-state geometry requirement for driving the reaction to the product according to the Lewis acid effect of lanthanides.15
To probe the heterogeneity of the catalysts, after the catalytic reactions, the catalysts were recovered from their reaction media. Solid of 3 could be easily isolated from the reaction suspension by a simple filtration and reused at least three times without an appreciable loss of its high catalytic performance (from 96.2% to 93.5% yield see Table S1†). The PXRD patterns of the retrieved catalyst 3 were identical with those of the fresh catalyst (shown in Fig. 5). The IR spectrum of the recovered compound 3 was also identical to that of the freshly prepared sample (Fig. S15†). These observations suggested that 3 is a true heterogeneous catalyst. In addition, infrared spectroscopy of the catalyst 3 impregnated with benzaldehyde exhibited one broad C–O stretch at 1692.4 cm−1. The v(C–O) stretch had a red shift of 10.4 cm−1 from 1702.8 cm−1 of the free benzaldehyde (Fig. S16†). This experiment unambiguously demonstrated the possible activation of the substrate by the Nd3+ cation as Lewis acid site in 3. Furthermore, the control experiment for cyanosilylation of benzaldehyde with K5NaMo6V2O26·4H2O precursor in a homogeneous manner gave 58.4% yield, which is far lower than that of compound 3 in the heterogeneous manner. The higher yield with compound 3 is possibly attributed to the synergistic effect of the Lewis acid (Nd3+ cation) and the Lewis base (surface oxygen atoms of the [Mo6V2O26]6− POM) that activate respectively aldehydes and TMSCN at the same time. The coexistence of activated coupling partners on the same POM molecules in which [Mo6V2O26]6− polyoxoanion activates the nucleophile TMSCN and Nd3+ cation is responsible for the activation of the electrophile aldehydes, results in the effective promotion of cyanosilylation, which is consistent with the research conclusion of Mizuno and co-workers.16,17 The possible mechanism for the cyanosilylation reaction in the case of compound 3 is shown in Scheme S1.†
 | ||
| Fig. 5 Powder X-ray diffraction (PXRD) patterns of 3: calculated pattern from crystal data (blue line); experimental pattern before catalysis (red line); recovered catalyst 3 after 3 catalytic runs of the cyanosilylation of benzaldehyde (black line). | ||
Conclusions
In summary, three bi-supporting structures based on β-[Mo6V2O26]6− anion and lanthanide cations, are reported, which represent the first examples of inorganic species constructed from molybdenum–vanadium POMs and lanthanides. These inorganic materials not only exhibit good photocatalytic activities for the degradation of organic dye RhB under the irradiation of UV light, but also act as efficient heterogeneous Lewis acid–base catalysts for cyanosilylation of various aldehyde compounds under solvent-free conditions. The successful isolation of these species certainly provokes researchers' interest to develop new heterogeneous catalytic materials containing molybdenum–vanadium POMs and lanthanides.Acknowledgements
The authors thank the National Natural Science Foundation of China (21371027, 21271037, 20901013) and Fundamental Research Funds for the Central Universities (DUT12LK01) for financial supports.References
- (a) V. Soghomonian, Q. Chen, R. C. Haushalter, J. Zubieta and C. J. O'Connor, Science, 1993, 259, 1596 CAS; (b) A. Müller, S. Q. N. Shah, H. Bögge and M. Schmidtmann, Nature, 1999, 397, 48 CrossRef PubMed; (c) C. L. Hill, Chem. Rev., 1998, 98, 1 CrossRef CAS PubMed.
- (a) D. L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105 RSC; (b) A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009 CrossRef CAS PubMed; (c) D. Y. Du, L. K. Yan, Z. M. Su, S. L. Li, Y. Q. Lan and E. B. Wang, Coord. Chem. Rev., 2013, 257, 702 CrossRef CAS PubMed.
- (a) J. Zhang, J. Hao, Y. G. Wei, F. P. Xiao, P. C. Yin and L. S. Wang, J. Am. Chem. Soc., 2010, 132, 14 CrossRef CAS PubMed; (b) S. T. Zheng, J. Zhang and G. Y. Yang, Angew. Chem., Int. Ed., 2008, 47, 3909 CrossRef CAS PubMed; (c) C. Y. Sun, S. X. Liu, D. D. Liang, K. Z. Shao, Y. H. Ren and Z. M. Su, J. Am. Chem. Soc., 2009, 131, 1883 CrossRef CAS PubMed.
- (a) U. Kortz, M. G. Savelieff, F. Y. A. Ghali, L. M. Khalil, S. A. Maalouf and D. I. Sinno, Angew. Chem., Int. Ed., 2002, 41, 4070 CrossRef CAS; (b) X. L. Wang, H. L. Hu, G. C. Liu, H. Y. Lin and A. X. Tian, Chem. Commun., 2010, 46, 6485 RSC; (c) X. Wang, M. M. Zhang, X. L. Hao, Y. H. Wang, Y. Wei, F. S. Liang, L. J. Xu and Y. G. Li, Cryst. Growth Des., 2013, 13, 3454 CrossRef CAS; (d) A. Seliverstovab and C. Streb, Chem. Commun., 2014, 50, 1827 RSC.
- (a) A. M. Khenkin, G. Leitus and R. Neumann, J. Am. Chem. Soc., 2010, 132, 11446 CrossRef CAS PubMed; (b) R. Neumann and A. M. Khenkin, Chem. Commun., 2006, 2529 RSC; (c) N. Laronze, C. Marchal-Roch, N. Guillou, F. X. Liu and G. Hervé, J. Catal., 2003, 220, 172 CrossRef CAS; (d) X. L. Chen, Y. Liu, H. Wang, M. J. Yuan, X. H. Wang and Y. G. Chen, RSC Adv., 2014, 4, 11232 RSC.
- (a) L. Lisnard, A. Dolbecq, P. Mialane, J. Marrot, E. Rivière, S. A. Borshch, S. Petit, V. Robert, C. Duboc, T. McCormac and F. Sécheressea, Dalton Trans., 2006, 5141 RSC; (b) Y. Q. Lan, S. L. Li, K. Z. Shao, X. L. Wang, X. R. Hao and Z. M. Su, Dalton Trans., 2009, 940 RSC; (c) L. N. Xiao, Y. Wang, Y. Chen, Y. Peng, G. D. Li, X. B. Cui, S. Y. Shi, T. G. Wang, Z. M. Gao and J. Q. Xu, Inorg. Chem. Commun., 2010, 13, 1217 CrossRef CAS PubMed; (d) W. L. Ge, Z. Y. Long, X. C. Cai, Q. Wang, Y. Zhou, Y. Xu and J. Wang, RSC Adv., 2014, 4, 45816 RSC.
- (a) J. X. Meng, Y. G. Li, H. Fu, X. L. Wang and E. B. Wang, CrystEngComm, 2011, 13, 649 RSC; (b) J. Y. Niu, G. Chen, J. W. Zhao, C. F. Yu, P. T. Ma and J. P. Wang, Cryst. Growth Des., 2010, 10, 4689 CrossRef CAS; (c) J. Fu, H. X. Sun, Y. Xu, C. L. Wang, D. R. Zhu, Q. Sun and H. K. Liu, CrystEngComm, 2012, 14, 5148 RSC; (d) Z. H. Shi, Y. S. Zhou, L. J. Zhang, C. C. Mu, H. Z. Ren, D. Hassan, D. Yang and H. M. Asifa, RSC Adv., 2014, 4, 50277 RSC.
- S. Y. Shi, Y. C. Zou, X. B. Cui, J. N. Xu, Y. Wang, G. W. Wang, G. D. Yang, J. Q. Xu, T. G. Wang and Z. M. Gao, CrystEngComm, 2010, 12, 2122 RSC.
- (a) C. M. Liu, D. Q. Zhang, M. Xiong and D. B. Zhu, Chem. Commun., 2002, 1416 RSC; (b) L. M. Duan, C. L. Pan, J. Q. Xu, X. B. Cui, F. T. Xie and T. G. Wang, Eur. J. Inorg. Chem., 2003, 2578 CrossRef CAS.
- (a) J. Q. Sha, J. Peng, H. S. Liu, J. Chen, A. X. Tian and P. P. Zhang, Inorg. Chem., 2007, 46, 11183 CrossRef CAS PubMed; (b) F. Y. Li, F. Z. Meng, L. F. Ma, L. Xu, Z. X. Sun and Q. Gao, Dalton Trans., 2013, 42, 12079 RSC.
- M. Šimuneková, D. Prodius, V. Mereacre, P. Schwendt, C. Turta, M. Bettinelli, A. Speghini, Y. H. Lan, C. E. Ansonb and A. K. Powell, RSC Adv., 2013, 3, 6299 RSC.
- (a) D. Liu, Y. Lu, H. Q. Tan, W. L. Chen, Z. M. Zhang, Y. G. Li and E. B. Wang, Chem. Commun., 2013, 49, 3673 RSC; (b) J. W. Zhao, D. Y. Shi, L. J. Chen, P. T. Ma, J. P. Wang, J. Zhang and J. Y. Niu, Cryst. Growth Des., 2013, 13, 4368 CrossRef CAS.
- (a) C. Boglio, G. Lemiére, B. Hasenknopf, S. Thorimbert, E. Lacôte and M. Malacria, Angew. Chem., Int. Ed., 2006, 45, 3324 CrossRef CAS PubMed; (b) N. Dupré, P. Rémy, K. Micoine, C. Boglio, S. Thorimbert, E. Lacôte, B. Hasenknopf and M. Malacria, Chem.–Eur. J., 2010, 16, 7256 CrossRef PubMed; (c) H. E. Moll, B. Nohra, P. Mialane, J. Marrot, N. Dupré, B. Riflade, M. Malacria, S. Thorimbert, B. Hasenknopf, E. Lacôte, P. A. Aparicio, X. López, J. M. Poblet and A. Dolbecq, Chem.–Eur. J., 2011, 17, 14129 CrossRef CAS PubMed.
- (a) X. Y. Liu, L. Wang, X. N. Yin and R. D. Huang, Eur. J. Inorg. Chem., 2013, 2181 CrossRef CAS; (b) X. L. Hao, M. F. Luo, X. Wang, X. J. Feng, Y. G. Li, Y. H. Wang and E. B. Wang, Inorg. Chem. Commun., 2011, 14, 1698 CrossRef CAS PubMed.
- (a) R. F. D'Vries, M. Iglesias, N. Snejko, E. Gutiérrez-Puebla and M. A. Monge, Inorg. Chem., 2012, 51, 11349 CrossRef PubMed; (b) R. F. D'Vries, V. A. Peña-O'Shea, N. Snejko, M. Iglesias, E. Gutiérrez-Puebla and M. A. Monge, Cryst. Growth Des., 2012, 12, 5535 CrossRef.
- Y. J. Kikukawa, K. Suzuki, M. Sugawa, T. Hirano, K. Kamata, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 3686 CrossRef CAS PubMed.
- K. Suzuki, M. Sugawa, Y. Kikukawa, K. Kamata, K. Yamaguchi and N. Mizuno, Inorg. Chem., 2012, 51, 6953 CrossRef CAS PubMed.
- Q. X. Han, X. P. Sun, J. Li, P. T. Ma and J. Y. Niu, Inorg. Chem., 2014, 53, 6107 CrossRef CAS PubMed.
- A. M. Nenner, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 1703 CrossRef.
- (a) G. M. Sheldrick, SHELXL 97, Program for Crystal Structure Refinement, University of Gőttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, SHELXL 97, Program for Crystal Structure Solution, University of Gőttingen, Germany, 1997 Search PubMed.
- (a) P. Mialane, L. Lisnard, A. Mallard, J. Marrot, E. Antic-Fidancev, P. Aschehoug, D. Vivien and F. Sécheresse, Inorg. Chem., 2003, 42, 2102 CrossRef CAS PubMed; (b) C. Zhang, R. C. Howell, K. B. Scotland, F. G. Perez, L. Todaro and L. C. Francesconi, Inorg. Chem., 2004, 43, 7691 CrossRef CAS PubMed; (c) L. B. Ni, F. Hussain, B. Spingler, S. Weyeneth and G. R. Patzke, Inorg. Chem., 2011, 50, 4944 CrossRef CAS PubMed.
- M. Sadakane, M. H. Dickman and M. T. Pope, Angew. Chem., Int. Ed., 2000, 39, 3036 CrossRef.
- H. Y. An, Z. B. Han and T. Q. Xu, Inorg. Chem., 2010, 49, 11403 CrossRef CAS PubMed.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244 CrossRef.
- A. M. Khenkin, P. Carl, D. Baute, A. M. Raitsimring, A. V. Astashkin, L. J. W. Shimon, D. Goldfarb and R. Neumann, Inorg. Chim. Acta, 2006, 359, 3072 CrossRef CAS PubMed.
- (a) K. Wang, D. D. Zhang, J. C. Ma, P. T. Ma, J. Y. Niu and J. P. Wang, CrystEngComm, 2012, 14, 3205 RSC; (b) J. Y. Niu, S. W. Zhang, H. N. Chen, J. Zhao, P. T. Ma and J. P. Wang, Cryst. Growth Des., 2011, 11, 3769 CrossRef CAS.
- (a) Y. H. Guo and C. W. Hu, J. Mol. Catal. A: Chem., 2007, 262, 136 CrossRef CAS PubMed; (b) T. H. Li, S. Y. Gao, F. Li and R. Cao, J. Colloid Interface Sci., 2009, 338, 500 CrossRef CAS PubMed.
- J. W. Sun, M. T. Li, J. Q. Sha, P. F. Yan, C. Wang, S. X. Li and Y. Pan, CrystEngComm, 2013, 15, 10584 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The coordination modes of La3+ cations in 1 and Nd3+ cation in 3, 3D supramolecular topology structure for 1, IR, TG curves and PXRD patterns for 1–3, UV-vis-NIR diffuse reflectance spectrum for K5NaMo6V2O26·4H2O, UV-vis absorption spectra of RhB solutions without catalyst and with K5NaMo6V2O26·4H2O as catalyst, effect of number of recycling cycles on the decolorization, kinetic profiles in the aldehyde cyanosilylation reaction for compounds 1–3, IR spectra for as-synthesized and recovered catalyst 3 are available. Study on recycling of catalyst 3 is listed in Table S1. See DOI: 10.1039/c4ra16237d |
| This journal is © The Royal Society of Chemistry 2015 |