
Fabrication of resin supported Au–Pd bimetallic nanoparticle composite to efficiently remove chloramphenicol from water
Xu Wangab, Yun-Xia Wangab, Baoling Yuanc, Hao-Jie Cuia and Ming-Lai Fu*a
aKey Laboratory of Urban Pollutant Conversion, Chinese Academy of Sciences, Xiamen 361021, China. E-mail: mlfu@iue.ac.cn; Fax: +86-592-6190762; Tel: +86-592-6190762
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cCollege of Civil Engineering, Huaqiao University, Xiamen, 361021, China
First published on 30th January 2015
Abstract
The evolution of antibiotic resistance and the potential impact on human health of chloramphenicol (CAP) have made it an environmental pollutant requiring urgent action. In this study, Au–Pd bimetallic nanoparticles (BNPs) were first synthesized and then successfully loaded on Amberlite 717 to form an Amberlite 717 supported Au–Pd BNP catalytic system (717@Au–Pd) with the mass fraction of Au–Pd at about 4.5%. The as-synthesized catalytic system was used to degrade CAP in water under a H2 atmosphere at room temperature. When 0.5 g of 717@Au–Pd was added into the CAP solution (50 mg L−1, 50 mL, pH 7), about 60% of CAP was absorbed on the 717@Au–Pd in the first 10 h and then all of CAP can be completely removed in the following 3 h under a H2 atmosphere. The degradation process of the reaction can be fitted with a first-order kinetics equation with the kinetics constant of 4.3 h−1 ± 0.009. The degradation products and mechanism were studied using LC/MSD Trap-XCT. The results showed that CAP was removed by the 717@Au–Pd via cleaving the carbon–halogen bond of CAP while keeping the nitro-group unaffected and this made the degradation products less environmentally toxic. The recycled experiments showed that the removal rate of CAP can still be maintained at 99% even after 5 cycles. The study indicated that 717@Au–Pd is a promising catalyst for removing environmental pollutants such as CAP containing carbon–halogen bonds.
Introduction
Chloramphenicol (CAP) is a broad-spectrum antibiotic exhibiting strong activity against most micro-organisms, including Gram-positive and Gram-negative bacteria. In the past, it has been widely used for the treatment of wounds and in ocular infections in clinical practice. In the last few decades, CAP has been prohibited in the treatment of food-producing animals in many countries, such as EU, USA, and Canada, because residual CAP in food may cause human bone marrow aplasia and therefore aplastic anemia.1 However, owing to its broad spectrum of action, high efficiency and relatively low cost, CAP is still in use illegally or legally in some countries. The persistent global use of CAP has led to its universal presence in the environment owing to its stability. For example, up to 26.6 μg L−1 of CAP has been found in effluents from sewage treatment plants in China.2 The persistent existence of CAP in the environment can result in the evolution of multidrug-resistant bacteria and novel antibiotic-resistant genes, which have attracted increasing attention because of their effects on human health.3,4 It is therefore urgent to search for effective methods to remove CAP in the environment.The molecule of CAP contains a benzene ring, two carbon–chlorine bonds and a nitro-group. The presence of carbon–chlorine bonds ensures it is a toxic chlorinated organic compound in the environment.5 Meanwhile, the nitro-group in the molecule of CAP is easily reduced to a nitroso-group or an amine group, which are strongly carcinogenic.6 In this context, it is an admirable objective to break the carbon–chlorine bonds and prevent the formation of nitroso- or amine groups during the degradation of CAP. In recent years, various methods have been used to degrade CAP. Among them, the advanced oxidation process (AOP) was considered to be an effective way to degrade CAP. Although many studies have already proved that the antimicrobial activity of CAP was negative after treatment with AOP methods, the degree of mineralization was still not high and the carbon–chlorine bonds may not be broken.2,7–10 Bioelectrochemical systems were also used for the degradation of CAP.11,12 In this method, the carbon–chlorine bonds could be broken, but the nitro-group could also be reduced to nitroso- or amine groups, an unfavorable outcome. Notably, promising nano-catalysts were also introduced to degrade CAP.13,14 For example, Singh et al. removed CAP by using zero-valent iron–silver bimetallic nanoparticles (ZVBMNPs) as catalysts,13 but the results showed that the by-products also contained an amine group, which was a probable precursor of N nitroso compounds. Furthermore, some other studies have suggested that CAP can be removed by reductive dechlorinating microorganisms or enzymes, but these studies showed that the process is very slow and still a long way from real application.15,16 The microwave radiation method was an effective method to remove CAP but the high radiation power made this method impossible in real applications.17 Therefore, it is important to find a promising way to cleave the carbon–chlorine bonds of CAP without reducing the nitro-group to amine or nitroso-groups.
Pd is a prominent catalyst that can cleave the carbon–chlorine bond in a H2 atmosphere,18 but the use of Pd as catalyst is limited as the catalytic efficiency of Pd is very low at room temperature. The catalytic performance of Pd catalyst can be enhanced when combined with other metals in various kinds of reaction.19–25 It has been shown that the catalytic performance of Pd in cleaving the carbon–halogen bond can be greatly improved by using Au–Pd bimetallic nanoparticles (BNPs).26,27 In our recent research, Au–Pd BNPs with a core–shell structure have also been proved to be an effective catalyst in the degradation of diclofenac by cleaving carbon–halogen bonds.28 As the performance of Pd catalyst in a H2 atmosphere has strict selectivity, it is possible to use Au–Pd BNPs to cleave carbon–chlorine bonds of CAP while keeping the nitro-group unaffected. Inspired by this idea, in this work, we used Au–Pd BNPs with a core–shell structure as catalyst to degrade CAP.
As the particle size of Au–Pd BNPs was too small to be separated and recycled easily, we utilized an anion exchange resin to support the Au–Pd BNP catalyst, with the aim of developing a simple and reusable catalytic system. First, Au–Pd BNPs with a core–shell structure were synthesized and then supported on the surface of a strong-base anion exchange resin (Amberlite 717) for the degradation of CAP. Amberlite 717 supported Au–Pd BNP composite was found to be an efficient catalyst for the dechloridation reaction of CAP, and the nitro-group remained unaffected during the reaction. This suggested composite could be used as a promising material for degradation of CAP in water without producing highly toxic intermediate substances.
Experimental
Preparation of Au NPs
Au NPs were synthesized according to a reported method.29 In a typical synthesis, a gold salt solution (125 mg L−1, 80 mL) was heated to 60 °C with stirring. Then a reducing agent with 40 mg of sodium citrate, 50 mg of tannic acid, and 18 mg of potassium carbonate dissolved in 20 mL of ultrapure water was added to the gold salt solution. The solution was then heated to boiling. Two minutes later, the solution was removed from the heat source and the Au NP solution (58 mg L−1, 100 mL) was obtained.Preparation of Au–Pd BNPs
Au–Pd BNPs were also synthesized according to a reported method.29 First, 2 mL of 600 mg L−1 H2PdCl4 solution, 10 mL of 58 mg L−1 Au NP solution and 10 mL of ultrapure water were mixed and stirred for more than 20 min. Then, H2 was bubbled through the solution for at least 3 min, and the Au–Pd BNP solution was obtained. By diluting the Au–Pd BNPs to a certain volume of ultrapure water, a 50 mg L−1 Au–Pd BNP solution with a mass ratio of Au/Pd 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was obtained.
:1 was obtained.Pretreatment of amberlite 717
Amberlite 717 (10 g) was first soaked in ethanol solution (50 mL) for 2 h, and then soaked sequentially in aqueous NaOH solution (2 M, 50 mL), and HCl solution (2 M, 50 mL) for several cycles, and finally in NaOH solution (4 M, 50 mL) for 4 h to exchange anions from Cl− to OH−. The resultant resin was filtered and washed with water until the filtrate was neutral. After that, the resin was dried at 80 °C overnight. The Amberlite 717 used in this study was pretreated with this procedure unless otherwise noted.Preparation of Amberlite 717 supported Au–Pd BNP catalytic system (717@Au–Pd)
Amberlite 717 (10 g) was soaked in Au–Pd BNP solution (50 mg L−1, 50 mL) for 1 h and then was separated. The resultant resin was washed with water and then dried at 80 °C. The above procedure was repeated several times until the resin attained saturation, which was indicated by the filtrate of washing water appearing obviously dark brown. Finally, the Amberlite 717 supported Au–Pd BNP catalytic system (717@Au–Pd) was obtained and used in this study.Characterization
A Hitachi S-4800 SEM and a JEOL JEM-2010 HRTEM were used to determine the structure of samples. A NETZSCH Tarsus TG 209 F3 was used for thermogravimetric analysis to evaluate the amount of Au–Pd BNPs loaded on the Amberlite 717. Sample masses of around 10 mg were heated from 40 to 1000 °C at a rate of 5 °C min−1 in a N2 atmosphere. An Agilent Technologies 1260 Infinity HPLC with an autosampler and a ZORBAX SB-C18 column (4.6 × 250 mm, 5 μm) was used to analyze the concentration change of CAP and its degradation products. The mobile phase was composed of 45%/55% (v/v) methanol and water at a flow rate of 1.0 mL min−1. The injection volume was 20 μL, and CAP and its degradation products were detected by an UV detector at a wavelength of 278 nm. CAP and its degradation products were monitored and identified using an Agilent Technologies 1100 LC/MSD Trap-XCT. Chromatography was carried out on a Nano HPLC comprising a quaternary pump and an autosampler, equipped with a ZORBAX Eclipse XDB-C18 column (4.6 × 150 mm, 5 μm). The mobile phase was composed of 45%/55% (v/v) methanol and water. Analytes were detected in negative ion mode. A PerkinElmer Optima 7000DV ICP-OES was used for analyzing concentrations of elemental Au and Pd released to the liquid phase.Catalytic performance testing
In a typical catalytic degradation process, the reaction was carried out in a 50 mL round-bottomed flask equipped with a magnetic stirrer. As CAP could be partly adsorbed by Amberlite 717, we performed a pretreatment process before the catalytic degradation started. 717@Au–Pd (0.5 g) was first mixed with 50 mL of CAP (50 mg L−1) and then stirred in the flask for 10 h to achieve adsorption equilibrium. Then the degradation reaction was carried out in a H2 atmosphere at pH 7 and at room temperature unless noted otherwise.Results and discussion
Characterization
The morphology and composition of 717@Au–Pd were characterized by SEM, EDAX and TEM. Fig. 1a shows a full view of 717@Au–Pd. The size of Amberlite was about 600 μm, which ensured it was an excellent macroscopic supporter in the catalytic system. In our previous research, it was already shown that the Au–Pd BNPs were of core–shell structure with Au as core and Pd as shell and the particle size of the BNPs was 7.88 ± 1.59 nm.28 After loading, as shown in Fig. 1b and c, Au–Pd BNPs were successfully supported on Amberlite 717. The BNPs were well distributed on the surface of Amberlite 717 and the sizes of BNPs were homogeneous at about 8 nm, which was similar to that in our previous study.28 As shown in Fig. 1d, EDAX spectra also confirmed the existence of Au and Pd in the composite. | ||
| Fig. 1 SEM images of 717@Au–Pd of low magnification (a), high magnification (b), HRTEM image of 717@Au–Pd (c), and EDAX spectra of 717@Au–Pd (d). | ||
In the literature, metallic/bimetallic nanoparticles have been supported on various carriers and used in the degradation of organic pollutants.30–32 As far as Au–Pd BNPs are concerned, the nanomaterials have been successfully supported on graphene oxide, metal oxides and many other macro supporters, and the catalytic activity of the resulting composites was always highly enhanced compared with that of the unsupported Au–Pd BNPs.33–40 But for Au–Pd BNPs with a core–shell structure, literature reports are rare. Edwards et al. loaded Au–Pd BNPs with a core–shell structure on carbon, TiO2 and Al2O3 using the incipient wetness method.41 The catalyst was used for the direct synthesis of hydrogen peroxide with high efficiency and selectivity. This composite was far less applicable in the treatment of water owing to difficult separation. Sarkany et al. loaded Au–Pd BNPs with a core–shell structure on SiO2 for acetylene hydrogenation. The catalytic activity of the catalyst decreased with increasing thickness of the Pd shell.25 In this study, by stepwise synthesis, Au–Pd BNPs with a core–shell structure were successfully supported on Amberlite by a simple method, which might be used to treat polluted water because of easy separation, low cost and high throughput.
In order to determine the amount of Au–Pd in the composite, thermogravimetry (TG) – derivative thermogravimetry (DTG) was used to analyze the decomposition of the composite. Fig. 2 shows the thermogravimetric analysis process of Amberlite 717 and 717@Au–Pd from 40 °C to 1000 °C. The residual mass percentages of Amberlite 717 and 717@Au–Pd were w1% = 12.91% and w2% = 16.80%, respectively.
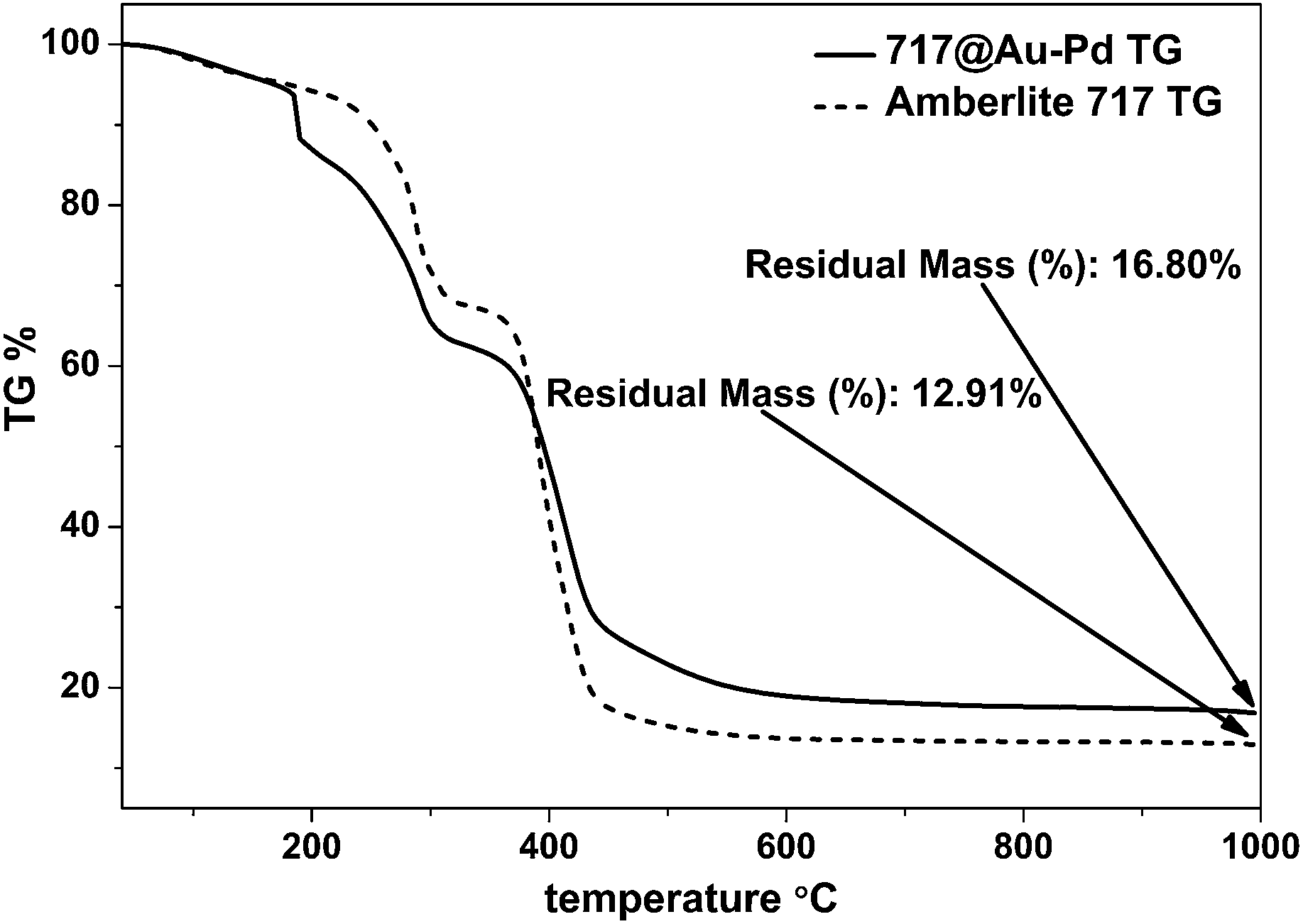 | ||
| Fig. 2 TG and DTG curves of Amberlite 717 and 717@Au–Pd. | ||
The residual mass percentage of Amberlite 717 can be described by eqn (1):
| w1% = w′a1/wa1 × 100% | (1) |
As with eqn (1), the residual mass percentage of 717@Au–Pd can be described by eqn (2):
 | (2) |
Then, the mass percentage of Au–Pd BNPs loaded on the Amberlite 717 (wBNPs%) can be described by eqn (3).
| wBNPs% = wBNPs/(wa2 + wBNPs) × 100% | (3) |
By combining these three equations, wBNPs% can be calculated by the following equation:
 | (4) |
The calculated mass percentage of Au–Pd BNPs successfully loaded on Amberlite 717 was about 4.5%.
Catalytic performance of 717@Au–Pd
It is well known that the catalytic performance of Pd in breaking carbon–halogen bonds can be greatly improved by forming Au–Pd BNPs with a core–shell structure.26 Meanwhile, it is necessary to support the Au–Pd BNPs on a macro substrate for development of a simple and reusable catalytic system as the particle size of Au–Pd BNPs is too small to be separated and recycled easily. In this study, a series of controlled experiments were set to evaluate the catalytic performance of 717@Au–Pd. As shown in Fig. 3, CAP can be partly adsorbed by Amberlite 717 and 717@Au–Pd. After pretreatment of 717 and 717@Au–Pd to the adsorption equilibrium, about 60% of CAP was absorbed before catalytic degradation started. For Amberlite 717, the concentration of CAP remained nearly constant in the following experimental process with or without a H2 atmosphere. This result showed that Amberlite 717 has good adsorption properties for CAP and it reached equilibrium after pretreatment with CAP for 10 hours. For the 717@Au–Pd composite, a similar amount (about 60%) of CAP was absorbed after the pretreatment process. In the following catalytic degradation, the concentration of CAP remained constant for 717@Au–Pd without a H2 atmosphere. In contrast, an obvious CAP concentration decrease was shown for 717@Au–Pd with a H2 atmosphere. After 3 h of reaction, no CAP could be detected by HPLC. The results showed that 717@Au–Pd has an excellent ability to remove CAP in a H2 atmosphere and the removal was caused by the synergistic effect of the catalytic degradation reaction by Au–Pd BNPs and the adsorption properties of Amberlite 717. | ||
| Fig. 3 Concentration variation of CAP under different experimental conditions, C0: 50 mg L−1. | ||
Curve 4 in Fig. 3 suggests that the catalytic degradation process of CAP may comply with an apparent first-order kinetics model. From kinetic analysis, the concentration data for CAP showed a good agreement with apparent first-order kinetics with the initial concentration of CAP at about 22 mg L−1, yielding an apparent rate constant (k1) of 4.3 h−1 ± 0.009 for 717@Au–Pd in a H2 atmosphere. Several groups have also studied the first-order reaction rate constant of CAP degradation reactions in various reaction systems.7–10,14 In most of these studies, the initial concentration of CAP was above 50 mg L−1 and they all degraded CAP with comparable first-order reaction rates, which suggested that these methods are promising ways to remove CAP. In this study, the removal of CAP was caused by the synergistic effects of adsorption following the degradation reaction. The first-order reaction rate constant can be used to describe the catalytic degradation process in the removal process. The high first-order reaction rate showed that 717@Au–Pd composite was a promising catalyst in degrading CAP.
In the literature, Wong et al. have been studying the hydrodechlorination property of Au–Pd BNPs with core–shell structure for a long time.19,26,42–45 In their studies, chloroform, trichloroethene, and perchloroethene were selected as the hydrodechlorination target objects and the hydrodechlorination efficiencies and catalytic kinetics of Au–Pd BNPs were investigated. The results showed that the hydrodechlorination efficiencies of Au–Pd BNPs with a core–shell structure were clearly improved compared with that of Pd catalyst. De Corte et al. also studied the hydrodechlorination property of biosupported Au–Pd BNPs for the removal of environmental contaminants such as trichloroethene and diclofenac.30,46 They confirmed that by combining Pd with Au, the catalytic activity of the catalyst can be greatly enhanced. Tanaka et al. prepared TiO2 loaded Au–Pd BNPs as catalysts and studied the dechlorination property to remove chlorobenzene under irradiation by visible light.47 The results showed that the conversion of chlorobenzene was improved by using Au(0.8)@Pd(0.2)/TiO2 instead of Pd(0.2)–TiO2. All of these studies unambiguously proved that Au–Pd BNPs was a promising catalyst in hydrodechlorination applications.
Mechanism of degradation
The degradation reaction was carried out with 717@Au–Pd in a H2 atmosphere at pH 7, and when the reaction ended, the pH of the solution was found to be about 5.5. It was already known that when the carbon–halogen bond is broken by Pd catalyst in a H2 atmosphere, the pH of the reaction system would be lowered as HCl is produced. Therefore, it can be inferred that the carbon–halogen bonds might be cleaved in this degradation reaction. Fig. 4a shows chromatograms of the degradation products of CAP at pH 7 using 717@Au–Pd in a stream of H2 bubbles after 45 min of reaction time. As shown, up to 6 peaks were observed and peak 1 belongs to that of CAP. The HPLC spectra implied that there may be more than 5 by-products present in the degradation process. A further study was carried out with an Agilent Technologies 1100 LC/MSD Trap-XCT. The LC-MS spectra showed that only CAP and two other by-products could be detected, as shown in Fig. 4b–d. | ||
| Fig. 4 HPLC and LC-MS of CAP and its by-products. (a) HPLC spectrum of CAP treated with 717@Au–Pd for 45 min, (b) LC-MS of CAP, (c) LC-MS of 2-chloro-N-(1,3-dihydroxy-1-(4-nitrophenyl)propan-2-yl)acetamide, (d) LC-MS of N-(1,3-dihydroxy-1-(4-nitrophenyl)propan-2-yl)acetamide. | ||
In Fig. 4b, m/z of 322.5 (M−, 100%), 258.5 (41), 193.5 (39) and 151.5 (16) belonged to the three characteristic fragment ion peaks of CAP.48 The molecular ion peak and fragment peak of Fig. 4c and d conformed to 2-chloro-N-(1,3-dihydroxy-1-(4-nitrophenyl)propan-2-yl)acetamide and N-(1,3-dihydroxy-1-(4-nitrophenyl)propan-2-yl)acetamide, respectively. These two substances can be ascribed to the products of CAP losing one Cl and two Cl atoms, respectively. These degradation products proved that a standard catalytic hydrogenolytic cleavage occurred in the degradation of CAP by 717@Au–Pd, and it is worth mentioning that the nitro-group cannot be reduced to a nitroso- or amine group.
The possible mechanism of the degradation reaction route of CAP is shown in Fig. 5. In the stepwise degradation route, Pd can break either of the two carbon–chlorine bonds of CAP, and in the following degradation, the remaining carbon–chlorine bond can be further cleaved. In the one-step route, both of these carbon–chlorine bonds can be cleaved at the same time. This suggested degradation route of CAP by 717@Au–Pd is similar to that of the degradation of diclofenac using Au–Pd BNPs with a core–shell structure in our previous work.28 The stepwise or one-step cleavage of the two carbon-chlorines of CAP was also found in the possible pathways of CAP reduction by ZVBMNPs.13 In that study, Singh et al. found that both of the carbon–halogen bonds and the nitro group in the CAP molecule could be reduced completely by a one-step degradation route, or the carbon–halogen bonds of CAP were cleaved by a stepwise degradation route, forming the same intermediate substance of 2-chloro-N-(1,3-dihydroxy-1-(4-nitrophenyl)propan-2-yl)acetamide. The most interesting thing is that a different intermediate substance of CAP was found for the one-step degradation route. In another degradation of CAP by microwave radiation, the CAP was cleaved into 4-nitrobenzoic acid and CH2Cl2 or CHCl3.17 In contrast, 717@Au–Pd may be a more promising catalyst for degrading CAP owing to cleavage of the carbon–halogen bonds and preservation of the nitro group.
 | ||
| Fig. 5 The reaction route of CAP in H2 environment with 717@Au–Pd. | ||
CAP is a chiral compound and its structural formula is shown in the inset in Fig. 4b. The enantiomorphs of the organic molecule have different polar properties despite having the same molecular weight. Bjorn et al. showed the HPLC peaks of two pairs of enantiomorphs of CAP molecule in different mobile phase situations.49 Combined with the data provided by HPLC and LC-MS, tiny amounts of enantiomorphs of CAP existed in the raw samples and its enantiomorphous by-products were also produced in the degradation process. By supposing this, peak 1 and 2 belonged to the enantiomorph of CAP, while peaks 3, 4 and 5, 6 represented the two enantiomorphs of the by-products of CAP.
Recycling performance
In order to test the recycling performance of 717@Au–Pd, the recycling experiment on the degradation reaction of CAP was carried out by soaking 717@Au–Pd in NaOH solution (2 M, 50 mL) for 1 h and washing with water until the filtrate was neutral before each cycle. The recycling performance of 717@Au–Pd is shown in Fig. 6. After 5 cycles, the removal rate of CAP could still be maintained at 99%. It can be observed that the degradation performance was maintained without a significant decline with the increase in number of cycles. No elemental Au or Pd could be detected in the reaction liquid phase throughout the recycling experiment according to analysis with ICP-OES. These results indicate that the loading of Au–Pd BNPs on Amberlite 717 is very stable for the application of removing CAP in water. | ||
| Fig. 6 Cycling tests of degradation reaction of CAP using 717@Au–Pd as catalyst, C0: 50 mg L−1. | ||
Conclusions
In this study, Au–Pd BNPs with a core–shell structure were synthesized and loaded on Amberlite 717 to form a macro catalyst system of 717@Au–Pd. The as-synthesized 717@Au–Pd proved to be a promising catalyst for the cleavage of carbon–chlorine bonds in the CAP molecule and with preservation of the nitro-group. The catalytic process could be described by apparent first-order kinetics and k1 = 4.3 h−1 ± 0.009. The recycling experiments showed that the catalytic performance of 717@Au–Pd was stable under experiment conditions. The relatively high reaction rate and its stable recycling performance make 717@Au–Pd a promising catalyst for removing environmental pollutants such as CAP containing carbon–halogen bonds.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 51478449), Xiamen Science & Technology Major Program (no. 3502Z20131018), the Natural Science Foundation of Fujian Province (no. 2013J01213), and the National High Technology Research and Development Program (“863” Program) of China (no. 2012AA062606).Notes and references
- A. A. Yunis, Am. J. Med., 1989, 87, N44–N48 Search PubMed.
- A. G. Trovo, V. A. B. de Paiva, A. E. H. Machado, C. A. de Oliveira and R. O. Santos, Sol. Energy, 2013, 97, 596–604 CrossRef CAS PubMed.
- X. X. Zhang, T. Zhang and H. Fang, Appl. Microbiol. Biotechnol., 2009, 82, 397–414 CrossRef CAS PubMed.
- C. P. Andam, G. P. Fournier and J. P. Gogarten, FEMS Microbiol. Rev., 2011, 35, 756–767 CrossRef CAS PubMed.
- T. Hirata, O. Nakasugi, M. Yoshioka and K. Sumi, Water Sci. Technol., 1992, 25, 9–16 CAS.
- R. Shen and S. A. Andrews, Water Res., 2011, 45, 944–952 CrossRef CAS PubMed.
- A. Zuorro, M. Fidaleo, M. Fidaleo and R. Lavecchia, J. Environ. Manage., 2014, 133, 302–308 CrossRef CAS PubMed.
- S. Garcia-Segura, E. B. Cavalcanti and E. Brillas, Appl. Catal., B, 2014, 144, 588–598 CrossRef CAS PubMed.
- M. H. Nie, Y. Yang, Z. J. Zhang, C. X. Yan, X. N. Wang, H. J. Li and W. B. Dong, Chem. Eng. J., 2014, 246, 373–382 CrossRef CAS PubMed.
- A. Chatzitakis, C. Berberidou, I. Paspaltsis, G. Kyriakou, T. Sklaviadis and I. Poulios, Water Res., 2008, 42, 386–394 CrossRef CAS PubMed.
- F. Sun, H. Liu, B. Liang, R. T. Song, Q. Yan and A. J. Wang, Bioresour. Technol., 2013, 143, 699–702 CrossRef CAS PubMed.
- D. Y. Kong, B. Liang, D. J. Lee, A. J. Wang and N. Q. Ren, J. Environ. Sci., 2014, 26, 1689–1697 CrossRef CAS PubMed.
- K. P. Singh, A. K. Singh, S. Gupta and P. Rai, Environ. Sci. Pollut. Res., 2012, 19, 2063–2078 CrossRef CAS PubMed.
- A. Jodat and A. Jodat, Desalin. Water Treat., 2014, 52, 2668–2677 CrossRef CAS.
- Y. B. Ahn, S. K. Rhee, D. E. Fennell, L. J. Kerkhof, U. Hentschel and M. M. Haggblom, Appl. Environ. Microbiol., 2003, 69, 4159–4166 CrossRef CAS.
- W. Tao, M. H. Lee, J. Wu, N. H. Kim, J. C. Kim, E. Chung, E. C. Hwang and S. W. Lee, Appl. Environ. Microbiol., 2012, 78, 6295–6301 CrossRef CAS PubMed.
- L. Lin, S. H. Yuan, J. Chen, L. L. Wang, J. Z. Wan and X. H. Lu, Chemosphere, 2010, 78, 66–71 CrossRef CAS PubMed.
- R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320–2322 CrossRef CAS.
- M. O. Nutt, J. B. Hughes and M. S. Wong, Environ. Sci. Technol., 2005, 39, 1346–1353 CrossRef CAS.
- H. Hildebrand, K. Mackenzie and F. D. Kopinke, Appl. Catal., B, 2009, 91, 389–396 CrossRef CAS PubMed.
- P. X. Wu, C. M. Liu, Z. J. Huang and W. M. Wang, RSC Adv., 2014, 4, 25580–25587 RSC.
- E. M. Zahran, D. Bhattacharyya and L. G. Bachas, J. Mater. Chem., 2011, 21, 10454–10462 RSC.
- M. S. Chen, D. Kumar, C. W. Yi and D. W. Goodman, Science, 2005, 310, 291–293 CrossRef CAS PubMed.
- E. Gross, I. Popov and M. Asscher, J. Phys. Chem. C, 2009, 113, 18341–18346 CAS.
- A. Sarkany, O. Geszti and G. Safran, Appl. Catal., A, 2008, 350, 157–163 CrossRef CAS PubMed.
- M. O. Nutt, K. N. Heck, P. Alvarez and M. S. Wong, Appl. Catal., B, 2006, 69, 115–125 CrossRef CAS PubMed.
- J. A. Rodriguez, Surf. Sci. Rep., 1996, 24, 225–287 CrossRef.
- X. Wang, J.-R. Li, M.-L. Fu, B. Yuan, H.-J. Cui and Y.-F. Wang, Environ. Technol., 2014, 1–25 CrossRef PubMed.
- J. W. Slot and H. J. Geuze, Eur. J. Cell Biol., 1985, 38, 87–93 CAS.
- S. De Corte, T. Hennebel, J. P. Fitts, T. Sabbe, V. Biznuk, S. Verschuere, D. van der Lelie, W. Verstraete and N. Boon, Environ. Sci. Technol., 2011, 45, 8506–8513 CrossRef CAS PubMed.
- M. El-Roz, Z. Haidar, L. Lakiss, J. Toufaily and F. Thibault-Starzyk, RSC Adv., 2013, 3, 3438–3445 RSC.
- L. Yang, L. Lv, S. J. Zhang, B. C. Pan and W. M. Zhang, Chem. Eng. J., 2011, 178, 161–167 CrossRef CAS PubMed.
- J. J. Lv, S. S. Li, A. J. Wang, L. P. Mei, J. R. Chen and J. J. Feng, Electrochim. Acta, 2014, 136, 521–528 CrossRef CAS PubMed.
- C. Ma, Y. Y. Du, J. T. Feng, X. Z. Cao, J. Yang and D. Q. Li, J. Catal., 2014, 317, 263–271 CrossRef CAS PubMed.
- A. Cybula, J. B. Priebe, M. M. Pohl, J. W. Sobczak, M. Schneider, A. Zielhiska-Jurek, A. Bruckner and A. Zaleska, Appl. Catal., B, 2014, 152, 202–211 CrossRef PubMed.
- F. Ruffino and M. G. Grimaldi, J. Nanopart. Res., 2011, 13, 2329–2341 CrossRef CAS.
- G. Hamm, C. Becker and C. R. Henry, Nanotechnology, 2006, 17, 1943–1947 CrossRef CAS.
- Z. Kaszkur, Phys. Chem. Chem. Phys., 2004, 6, 193–199 RSC.
- S. N. Reifsnyder and H. H. Lamb, J. Phys. Chem. B, 1999, 103, 321–329 CrossRef CAS.
- A. A. Schmidt and R. Anton, Surf. Sci., 1995, 322, 307–324 CrossRef CAS.
- J. K. Edwards, S. J. Freakley, A. F. Carley, C. J. Kiely and G. J. Hutchings, Acc. Chem. Res., 2014, 47, 845–854 CrossRef CAS PubMed.
- J. C. Velazquez, S. Leekumjorn, Q. X. Nguyen, Y. L. Fang, K. N. Heck, G. D. Hopkins, M. Reinhard and M. S. Wong, AlChE J., 2013, 59, 4474–4482 CrossRef CAS.
- S. J. Li, Y. L. Fang, C. D. Romanczuk, Z. H. Jin, T. L. Li and M. S. Wong, Appl. Catal., B, 2012, 125, 95–102 CrossRef CAS PubMed.
- Z. Zhao, Y. L. Fang, P. J. J. Alvarez and M. S. Wong, Appl. Catal., B, 2013, 140, 468–477 CrossRef PubMed.
- Y. L. Fang, K. N. Heck, P. J. J. Alvarez and M. S. Wong, ACS Catal., 2011, 1, 128–138 CrossRef CAS.
- S. De Corte, T. Sabbe, T. Hennebel, L. Vanhaecke, B. De Gusseme, W. Verstraete and N. Boon, Water Res., 2012, 46, 2718–2726 CrossRef CAS PubMed.
- A. Tanaka, K. Fuku, T. Nishi, K. Hashimoto and H. Kominami, J. Phys. Chem. C, 2013, 117, 16983–16989 CAS.
- H. T. Ronning, K. Einarsen and T. N. Asp, J. Chromatogr. A, 2006, 1118, 226–233 CrossRef CAS PubMed.
- B. J. A. Berendsen, M. L. Essers, L. A. M. Stolker and M. W. F. Nielen, J. Chromatogr. A, 2011, 1218, 7331–7340 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |