
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic degradation and toxicity evaluation of diclofenac by nanotubular titanium dioxide–PES membrane in a static and continuous setup
K.
Fischer
a,
M.
Kühnert
a,
R.
Gläser
b and
A.
Schulze
*a
aLeibniz Institute of Surface Modification, Permoserstraße 15, Leipzig, D-04318, Germany. E-mail: agnes.schulze@iom-leipzig.de; Tel: +49 341 235 2400
bLeipzig University, Institute of Chemical Technology, Linnéstraße 3, Leipzig, D-04103, Germany
First published on 19th January 2015
Abstract
Diclofenac is a commonly used anti-inflammatory drug, which has been found in surface waters. Advanced oxidation processes (AOPs) seem to be the most suitable technique to prevent the entry of diclofenac and other pollutants into surface waters. TiO2 is especially reliable in mineralizing many organic molecules. The combination of TiO2 nanotubes with a polymer microfiltration membrane (polyethersulfone, PES) showed high photocatalytic activity by degrading diclofenac combined with an excellent membrane performance and long-term stability. By continuously degrading pollutants from water via a cross-flow setup, the molecules to be degraded are transported right to the membrane surface so that the overall reaction rate is increased. The toxicity of diclofenac was reduced by photocatalysis and photolysis; however, photocatalysis had greater impact. Moreover, the complete degradation of pollutants is very important to avoid highly toxic intermediate products.
1. Introduction
Water is essential for all living forms on the earth. Access to clean and fresh water is especially important and has therefore been declared by the United Nations General Assembly as a human right (Resolution 64/292). The industrialization and growth of the human population in the last centuries has led to an increasing demand for clean water, but simultaneously more and more pollutants have entered the water cycle. Waste water treatment plants (WWTPs) do clean the water from many pollutants, but the increasing entry of emerging contaminants (e.g. pharmaceuticals, personal care products, steroid hormones, industrial chemicals, and pesticides) was not properly considered when designing today's WWTPs and do pass through the cleaning process due to their persistency and/or their continuous introduction.1 Therefore, many of these compounds enter the water cycle and harm wildlife and human life. The increasing identification of many pharmaceuticals in surface and drinking water has led to an increased concern and the investigation of the occurrence, fate and removal of pharmaceuticals from water.2,3 More than 150 drugs have been found in waste, surface and ground water.4 Diclofenac is one of the most often detected pharmaceuticals in the environment at concentrations in the surface water ranging from 0.14 to 1.48 μg L−1.5 As a non-steroidal anti-inflammatory drug, it reduces inflammation and relieves pain in patients. Because diclofenac is one of the most used painkillers and WWTPs do not completely eliminate it from the waste water (<0 to 81.4%),1 it was one of the first pharmaceuticals found in the aquatic environment.6 Therefore, in 2013 diclofenac and two estrogenic hormones were included in the watch list of the European Union (Directive 2013/39/EU).To successfully remove diclofenac from wastewater, Vieno et al.7 suggested a tertiary treatment step in WWTPs. Advanced oxidation processes (AOPs) show a high efficiency in cleaning recalcitrant wastewater8–10 and could be an ideal tool to completely remove diclofenac. AOPs generate strongly oxidizing hydroxyl radicals (OH˙) that mineralize organic molecules.11 The AOP technique UV/TiO2 has the additional advantage of mineralizing a wide range of organic compounds to water and carbon dioxide at ambient temperature and pressure. Contrary to other AOPs, however, it does not need activation by hydrogen peroxide or termination by quenching agents.8–12
TiO2 can be suspended in a reaction mixture (Type A reactor)13,14 or it can be fixed to a support material (Type B reactor, e.g. quartz, membrane).15–18 Suspended TiO2 has a larger surface area compared to the attached TiO2, but the TiO2 has to be filtered and re-suspended from the water, which is expensive and time consuming. Type B reactors can be used in a one-step process but the photocatalytic activity is decreased due to the loss of overall surface area associated with immobilizing the TiO2. The design of smart TiO2 morphologies (e.g. nanotubes, nanospheres, fibers) increases the photocatalytic activity and makes type B reactors attractive and effective. TiO2 nanotubes are particularly expected to be the promising structural design used to overcome the drawbacks because of their high surface-area-to-volume ratio, short distance for charge carrier diffusion and high photon-collection efficiency.19–22 Because membrane systems are often applied to purify water and to separate TiO2 particles in Type A reactors from cleaned water, they could even support TiO2 itself.18,23–26 Thus, the degradation of pollutants via TiO2 could be performed in a one-step process.
Recently, we have shown the synthesis via anodization of TiO2 nanotubes on an organic membrane (polyethersulfone, PES).26 The TiO2 nanotubes were strongly attached to the membrane and showed high photocatalytic activity in degrading methylene blue. In this study, we show the photocatalytic ability of nanotubular TiO2–PES membranes to degrade diclofenac not only in a stationary setup, but also in a cross-flow mode. In a cross-flow setup the molecules to be degraded (diclofenac) are transported directly to the reaction location, where OH radicals are produced. The stability of the nanotubular TiO2–PES membrane was tested and a change in permeation flux was observed. The change in water contact angle was monitored and was due to the effect of crystallization of the TiO2 nanotubes. The toxicity of intermediate products during photocatalysis and photolysis was tested by examining the viability of yeast cells (Saccharomyces cerevisiae). Yeast is a suitable test organism to monitor the toxicity of substances in water with a high sensitivity comparable to standard test systems like Daphnia magna and Vibrio fisheri.27–31
2. Experimental
2.1. Materials
Polyethersulfone membrane (Millipore Express® PLUS Membrane Filters, HPWP14250, pore size 0.45 μm) was obtained from Merck Millipore. Ethylene glycol (≥99%, for synthesis) was purchased from Carl Roth GmbH + Co. KG. Ammonium fluoride (for analysis EMSURE® ACS) was received from Merck KGaA. Palladium foil (99.9%, 1 mm thick), palladium wire (99.9%, 1.0 mm diameter) and titanium foil (99.6%, 0.05 mm thick) were purchased from ChemPur Feinchemikalien und Forschungsbedarf GmbH. Deionized water was obtained from a Milli-Q water filtration station (Millipore). Diclofenac sodium salt was received from the Cayman chemical company (≥99%). Baker's yeast (hydrated; Saccharomyces cerevisiae) was purchased from Fala GmbH, while fluorescein diacetate and D(+)-glucose anhydrous was obtained from Sigma Aldrich.2.2. Synthesis of TiO2 nanotubes
The synthesis of TiO2 nanotubes on a silicon wafer (for contact angle measurements) and PES membrane has been described elsewhere.26 Briefly, the PES membrane was first sputtered with titanium (thickness of 900 nm; Leybold Z 400). The electrolyte solution (ethylene glycol, ammonium fluoride 1.3 wt% and water 2 vol%) was aged via the anodization of titanium before the actual anodization of the membrane. The membrane was anodized by placing it in a two-electrode configuration, with a constant voltage of 30 V. The formation of TiO2 nanotubes on the PES membrane needed further modification to generate TiO2 nanotubes on a larger membrane area (60 × 70 mm). In contrast to the previously used method,26 the part of the membrane that is at the border of air and electrolyte was cooled with an iced metal block (sealed with parafilm), to avoid the generation of high temperatures. The membrane was kept in the electrolyte solution after termination of the anodization for up to 30 min. The anodized support was rinsed with deionized water and washed twice for 15 min with deionized water before drying in air.TiO2 was crystallized with water at low temperatures, and with water vapor at 110 °C for 1 h.26
2.3. Characterization
The morphology of the TiO2 nanotubes was characterized using a field emission scanning electron microscope (FESEM; Zeiss Ultra 55).The water permeation flux was examined with a stainless steel pressure filter holder (16249, Sartorius, Germany) for dead-end filtration. The membrane (active area: 17.35 cm2) was compacted with 0.5 bar and ultrapure water (100 mL). Time was monitored and the water permeation flux was calculated with the following equation:
 | (1) |
The contact angle of the TiO2 nanotubes was measured on Si wafers to obviate the capillary forces of the membrane. The commonly used method to press the membrane to a flat film32 in order to avoid capillary forces would destroy the TiO2 nanotubes. The static water contact angle was measured with the sessile drop method on a DSA II (Krüss, Germany). A drop (5 μL) was placed onto the Si wafers with a micro syringe.
The binding of TiO2 nanotubes to the surface was analyzed by rinsing the membrane surface with water. The membrane was fixed via double-sided tape to the inside of a glass beaker (80 mL), and 50 mL of water was added. The water was magnetically stirred at a high rate (1400 rpm) for 1 h so that a high force was established against the nanotubes. The surface of the membrane was examined via SEM before and after stirring.
The photoactivity of the TiO2 nanotubes on the PES membrane was tested in a static and a cross-flow experiment.
For the static experiment, the membranes were cut into circles (25 mm diameter) with a center hole (10 mm diameter, for comparability to experiments with methylene blue26). The cut membranes were secured to 6-well plates (ThermoFischer Scientific) with double-sided tape. Diclofenac sodium salt (25 mg L−1 or 5 mg L−1, 4 mL) was added to the well (ca. 1 cm height of solution in the well) with the membrane and the well plate was shaken in dark for 30 min for complete adsorption to take place. The mass of the plate was recorded to even out the loss of water during photocatalysis due to evaporation. The well plates were irradiated with a UV-A sunlamp (Heraus Original Hanau Suncare tanning tube 21/25 Slim, radiant flux density 7.6 mW cm−2). After certain time intervals the loss of water was valued by adding water to the well until the initial mass was reached and 60 μL (25 mg L−1 diclofenac) or 350 μL (5 mg L−1 diclofenac) of the solution was transferred to a UV-light transparent 96-well plate (UV-Star microplate, Greiner Bio-One) to measure the absorption at 276 nm using an Infinite M2 UV-Vis spectrophotometer (Tecan Group Ltd; Switzerland). The solution was transferred back to the 6-well plate and the plate was placed again below the UV-A sunlamp. Photolysis was measured with the same method excluding the membrane from the well. The rate constant was evaluated via first order kinetics. The slope of logarithmic c/c0 (where c is the concentration of diclofenac at time x of the photocatalytic degradation and c0 is the initial concentration of diclofenac) over time is the rate constant.
The cross-flow experiment was operated with a self-built setup that is schematically shown in Fig. 1. The membranes were cut into circles (47 mm diameter) and placed onto the sample holder of the cross-flow apparatus. Diclofenac solution (25 mg L−1 or 5 mg L−1, 80 mL) was added to the flask. Firstly, the system was operated for 30 min without UVA-light for complete dark adsorption to take place. The vacuum pump was set to 800 mbar and the membrane pump was started. The solution circulated from the flask to the membrane and through the membrane back to the flask. The inflow of the membrane pump was regulated by monitoring the liquid overlap (ca. 1 cm) on the membrane with a laser. By turning on the UV-LED lamp (6 UV-LED lights arranged in a circle, 365 nm, 7.5 mW cm−2, LZ1_00U600, LedEngin Inc.) the photocatalytic degradation was started. The decline in absorption at 276 nm was measured periodically in the spectrometer by transferring 60 μL (25 mg L−1 diclofenac) or 350 μL (5 mg L−1 diclofenac) from the flask to a 96-well plate (UV-Star microplate, Greiner Bio-One). The solution was transferred back to the flask after measuring the absorption. The total organic carbon (TOC) value was measured after 5 days and 10 days for the 25 mg L−1 diclofenac solution (the TOC value of the 5 mg L−1 diclofenac solution was below the detection limit). 50 mL of the sample was transferred to a flask by the liquid TOCII (elementar Analysensystem GmbH) and the TOC value of each sample was measured three times.
 | ||
| Fig. 1 Schematic of the cross-flow setup to measure the photocatalytic activity of the membrane in a flow-through mode. | ||
2.4. Evaluation of toxicity
The toxicity of untreated and treated (photocatalysis and photolysis) diclofenac was evaluated by monitoring the viability of Saccharomyces cerevisiae (baker's yeast).The yeast was stored at 10 °C. For cultivation, 1 mg of yeast and 30 mg of D(+)-glucose were suspended with 500 μL of treated (photocatalytically or photolytically) or untreated diclofenac solution to a 48-well plate. A water-yeast suspension without any diclofenac was prepared as a control sample (reference). The culture was incubated at 37 °C and was shaken for 4 h.
To measure the viability, the culture was treated with fluorescein diacetate (FDA). As FDA is membrane-permeable it can penetrate into yeast cells and is cleaved by the intracellular esterase of living cells to fluorescein.33 Fluorescein can be detected by fluorescence measurements (excitation 488 nm, emission 543 nm). Thus, a 0.2 mg L−1 solution was prepared by diluting a stock solution of FDA (5 mg L−1 in acetone, stored at −20 °C) in water right before adding it to each well (100 μL). The well plate was covered with aluminum foil and shaken for 10 min at room temperature. The amount of viable cells was measured via a spectrometer.
3. Results and discussion
3.1. Assembly and crystallization of TiO2 nanotubes on the membrane
The formation of TiO2 nanotubes on a PES membrane has been described elsewhere26 thoroughly. Usually the upscaling of the anodization to a larger area34 causes no problem as the titanium support can conduct large currents without causing any damage. Upscaling is not addressed in many publications as the focus has been directed only on the length35–38 and on the geometrical features39–41 of the nanotubes. In general, the temperature increases when the anodized area is enlarged due to the elevated flow of current, oxidation and etching during anodization. The electrolyte solution can remove the heat efficiently. However, at the interface of air and electrolyte solution (see Fig. 2), liquid was drawn up at the membrane. Therefore, anodization was also taking place at the part of the titanium-membrane-anode, which was not in direct contact with the electrolyte solution. The material heated up in this part was due to air being a poor heat conductor. By enlarging the anodized area the overall current increased, which led to an elevation in heat production. The PES membrane (glass transition temperature of 204 °C (ref. 42)) could not withstand such high temperatures leading to current breakdown at an early stage of the anodization. A different experimental setup to avoid the generation of heat (titanium support contacted and pressed against an O-ring inside the electrolyte solution)35 was not possible because the membrane material is insulating and permeable. The generation of TiO2 nanotubes on other support materials mostly uses high-temperature resistant materials like metals, glass, wafer, ITO, clay or Kapton.37,43–54 Only Galstyan et al.47 and Nanjo et al.46 used low-temperature resistant polymers, namely PET (polyethylene terephthalate) and PEN (polyethylene naphthalate), respectively. PEN can resist higher temperatures than PET (glass transition temperature of 76 °C (ref. 55)).56 Both the polymers were additionally covered with a film of ITO. Neither Galstyan et al.47 nor Nanjo et al.46 described any of the problems arising due to the generation of heat, and the sample dimensions also was not specified. With small sample dimensions less heat will be generated and the heat-factor can be neglected (as it was also experienced in the previous study26). | ||
| Fig. 2 Schematic illustration of the experimental setup for anodization of the PES membrane coated with titanium (Ti-membrane-anode). Electrolyte was soaked up at the part of the anode above the interface leading to increased temperature and a higher anodization rate. | ||
To avoid the high temperature, an iced metal block was pressed against the membrane in the interface region so that the heat was removed and the anodization of titanium to TiO2 nanotubes on the membrane samples (with an area of 60 × 70 mm) was possible (Fig. 3).
 | ||
| Fig. 3 FESEM images of TiO2 nanotube arrays on top of a PES membrane. The TiO2 nanotubes in (a) and (b) were amorphous while the nanotubes in (c) were crystallized to anatase with vapor-water at 110 °C. Image (b) is a magnification of image (a). | ||
The TiO2 nanotubes were crystallized via vapor-water to gain photoactive nanotubular TiO2 on top of the membrane (Fig. 3c).26
3.2. Water permeation flux
Due to the addition of 900 nm titanium on the membrane surface, the pore size of the membrane decreased (Fig. 4); moreover, the water permeation flux was reduced from 122 mL min−1 cm−2 bar−1 to 88 mL min−1 cm−2 bar−1 (Fig. 5). Adding such a thick film to a microfiltration membrane will change its membrane performance. The normally used 0.22 μm PES membrane was completely occluded when a 900 nm film of titanium was sputtered to it.26 Therefore, the permeation flux of the coated membrane showed a good value when comparing the water permeation flux of the unmodified 0.22 μm membrane with a value of 32 mL min−1 cm−2 bar−1 with the permeation flux of the titanium coated 0.45 μm membrane of 88 mL min−1 cm−2 bar−1. The anodization and crystallization did not change the permeation flux leading to a good value of 88 mL min−1 cm−2 bar−1 for the end product. | ||
| Fig. 4 FESEM images of the PES membrane with a pore size of 0.45 μm (a) and sputtered with a film of 900 nm titanium (b). | ||
 | ||
| Fig. 5 The water permeation flux of untreated membranes, membranes with titanium (900 nm) and membranes with TiO2 nanotubes (NT; amorphous and anatase) is shown. | ||
3.3. Water contact angle
The water contact angle was measured on a Si wafer to just gain the water contact angle values of the TiO2 nanotubes and not of the membrane substructure. The measurement of water contact angles on hydrophilic microfiltration membranes with the sessile drop method is challenging due to the capillary forces of the membrane. Usually the membrane needs to be pressed in order to measure a proper value.32 However, if the membrane with TiO2 nanotubes was pressed, the TiO2 nanotubes would be destroyed. The structuring of the titanium film (with a thin natural TiO2 layer on top) to form amorphous TiO2 nanotubes increased the water contact angle from 62° to a hydrophobic surface of 105° (Fig. 6). In general, an increase in roughness results in either an increase or decrease of the water contact angle. For TiO2 nanotubes, the water contact angle usually drops.57,58 Yoriya et al.59 have shown that the water contact angle differs between TiO2 nanotubes with different tube spacing. Large tube to tube spacing leads to higher contact angles, while densely packed TiO2 nanotube arrays generate lower contact angles. The nanotubes formed here have a high tube to tube spacing (Fig. 3b), which can be the reason for the hydrophobic properties. The crystallization of the TiO2 nanotubes led to the hydrophilization of the surface with a contact angle of 25°. This effect has been described in previously reported studies57–60 and can be explained by the effect of decreasing the tube to tube spacing.59 Due to the crystallization, the tube walls did grow and tube spacing was lost (Fig. 3c), which led to a decrease in the water contact angle.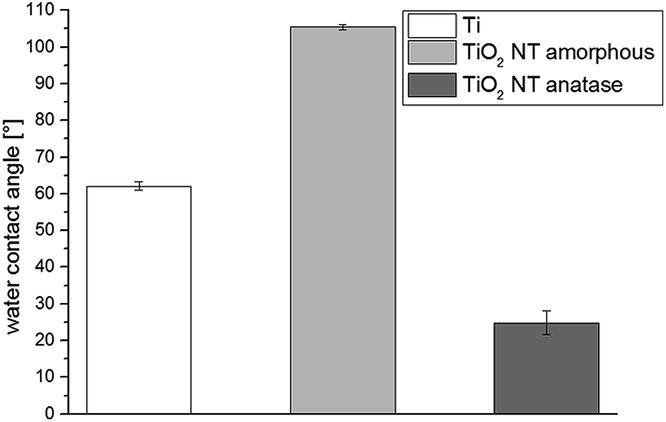 | ||
| Fig. 6 The water contact angle of Si wafer covered with a layer of titanium (Ti), with amorphous TiO2 nanotubes (TiO2 NT amorphous) and with crystallized anatase TiO2 nanotubes (TiO2 NT anatase) is displayed. | ||
3.4. Binding performance of the TiO2 nanotubes
The TiO2 nanotubes on the PES membrane were tested for their stability by applying a horizontal force executed by a continuous stream of water. The special morphology of the nanotubes on the membrane (Fig. 3) exhibited many attacking sides due the porous structure of the membrane and the nanotubes being aligned into different directions. The TiO2 nanotubes did not break up in detectable dimensions from the membrane (Fig. 7e and f) nor show significant change in the structure or loss of nanotubes, when examining the surface at a higher magnification (Fig. 7b and d). Up to five different spots on the membrane were surveyed, none of which showed any significant difference from the images displayed here. Breakage was expected, especially at the creases (due to the substructure of the membrane), but only one spot was found, where TiO2 nanotubes were detached from the surface. This spot is most likely not because of the detachment due to the water stream, but could be from the handling of tweezers. | ||
| Fig. 7 FESEM and photographical images of the TiO2 nanotubes on the PES membrane (as-anodized (a, b and e) and crystallized (c, d and f)) before and after the stability test. The TiO2 nanotubes on the membrane were tested on their stability by applying a horizontal force of a water stream to the membrane surface (b, d, e, and f). | ||
FESEM images of the nanotubular TiO2–PES membrane exposed to 10 days of cross-flow with water (800 mbar) also showed no breaking of the TiO2 nanotubes (Fig. 8) from the membrane surface.
 | ||
| Fig. 8 FESEM images of crystallized TiO2 nanotubes on the membrane after 10 days of cross-flow experiment at 800 mbar. The membrane surface does show some regions of organic fouling. Images (b) and (c) are magnifications of image (a). | ||
3.5. Degradation of diclofenac
The photocatalytic activity of the nanotubular TiO2–PES membrane has been shown recently in degrading the well-studied dye methylene blue.26 In Fig. 9(a) the degradation of diclofenac is shown for the static setup. The photocatalysis of diclofenac proceeds via hydroxyl derivative generation, ring opening and final mineralization.61 In the beginning (first 60 min) the degradation of a diclofenac solution with an initial concentration of 25 mg L−1 was quite similar to the degradation rate of a diclofenac solution with an initial concentration of 5 mg L−1 (Fig. 9(a) and Table 1). But after 60 min of photocatalytic treatment, the degradation rate slowed down for the diclofenac solution with an initial concentration of 25 mg L−1. This is also demonstrated when calculating the rate constant in the range of 60 to 240 min (Table 1), in which the value decrease by a factor of more than four. 94% of the diclofenac solution with an initial concentration of 5 mg L−1 was degraded after 240 min. The photolysis of diclofenac was slow and did not reach values below 30% of degraded diclofenac after 240 min. The rate constant is comparable to the values determined by Méndez-Arriaga et al. and Rizzo et al. (1 × 10−2 to 1 × 10−3 min−1),62,63 and it depended strongly on the loading of catalyst and concentration of diclofenac. The degradation rate increased with a lower initial concentration of diclofenac63,64 (Fig. 9(a) and Table 1). | ||
| Fig. 9 (a) The degradation of diclofenac was followed via spectrometry (initial concentration of 5 mg L−1 and 25 mg L−1) in the static (a) and cross-flow (b) setup. A TOC value was generated for the intermediate and end product of the photocatalysis and the end product of the photolysis of the diclofenac solution with an initial concentration of 25 mg L−1 in (b). The degradation progress in (b) was fitted via single exponential decay fitting (y = y0 + aekx, y0 is the value at the start, a is the amplitude and k the rate of decay) executed with OriginPro 9 g software. | ||
| Concentration of diclofenac [mg L−1] | Rate constant [min−1] static experiment | Rate constant [min−1] cross-flow experiment |
|---|---|---|
| a The rate constant was determined from 0 to 60 min and 60 to 240 min as the degradation slowed down after 60 min of degradation for the 25 mg L−1 diclofenac solution. | ||
| 5 | 9.96 ± 0.51 × 10−3 | 0.085 × 10−3 |
| 25 | 8.29 ± 1.25 × 10−3 (0–60 min)a | 0.057 × 10−3 |
| 25 | 1.92 ± 0.42 × 10−3 (60–240 min)a | |
The degradation of diclofenac in a continuous way (cross-flow) for the two different concentrations is shown in Fig. 9(b). Within the first three days of photocatalysis, the degradation did not vary between the diclofenac solutions with different concentrations. After 4 days the degradation of the 5 mg L−1 diclofenac solution increased (42% and 28% of diclofenac are degraded for 5 mg L−1 and 25 mg L−1, respectively). After 18 days of photocatalysis, the diclofenac solution with an initial concentration of 5 mg L−1 was degraded 100%, while the degradation of the diclofenac solution with an initial concentration of 25 mg L−1 needed to be continued further to gain 100% removal (estimated via a single exponential decay fitting). The photocatalytic conversion using the cross-flow setup also followed the general rule that with a lower initial concentration, the degradation rate is increased as it has also been found in the static experiment and is described in previous reports.64 The photolysis (demonstrated by repeating the cross-flow experiment with membranes with amorphous TiO2 nanotubes) of diclofenac was slow and did not reach values below 35% of degraded diclofenac after 10 days. The photolytic degradation of the diclofenac solution with 25 mg L−1 initial concentration was higher compared to diclofenac solution of 5 mg L−1 initial concentration. The photolysis of the solution with the higher initial concentration could have been affected by the precipitation of diclofenac at low pH values. Diclofenac might be not degraded but instead was adsorbed at the membrane in form of precipitates. Diclofenac does precipitate during photolysis due to the formation of hydrochloric and carboxylic acids and a resulting pH drop was observed.65 The solubility of diclofenac was reduced with decreasing pH.66,67 The photolysis of the diclofenac solution with a lower initial concentration might not have been affected by precipitation as the generation of hydrochloric and carboxylic acids was limited with lower concentration and/or the solubility at 5 mg L−1 was still maintained.
The TOC value matched the value generated by the spectrometric analysis of the diclofenac solution after degradation to a high degree. The obtained TOC values were below the spectrometric values. Mineralization should be slower,61 but taking the value at a progressive stage in the degradation (intermediates might be already degraded as well), having the eventual intermediates also absorbing at 276 nm and/or intermediates that are strongly bound to the catalyst might be the reason for the low TOC values.64 Another reason could be that diclofenac and its transformation products might also be further transformed by solar radiation68 during the TOC measurement. Spectrometric analysis was carried out immediately after sample withdrawal, whereas the actual measurement of the sample in the TOC device took up to 4 h. The evaluation of TOC values for the diclofenac solution with an initial concentration of 5 mg L−1 were not reasonable as the values for the degraded product were found to be below the detection limit.
The degradation rate of the cross-flow experiment was low when compared with the static experiment that had degradation rates about 100 times higher (Table 1). A direct comparison between these two experimental setups is not reasonable due to the difference in the membrane area, volume of diclofenac solution and amount of solution being non-irradiated in the flask and tubes during the cross-flow experiment. The membrane area (amount of catalyst) and the volume (water with diclofenac) increased from the static to the cross-flow experiment (4.1 cm2 to 12.6 cm2 and 4 mL to 80 mL). In addition, only 20% of the solution was irradiated at a time in the cross-flow setup leaving 80% unused in the flask and tubes. In order to yield higher degradation rates, the experimental setup of the cross-flow experiment needs to be optimized.
There was some fouling visible on the membrane after 10 days of the cross-flow experiment (Fig. 8). TiO2 is a well-known anti-fouling agent,18,69,70 which reduces the fouling of organic membranes, but with 10 days of constant water flow through the membrane and the fact that only the top part of the membrane was coated with TiO2 nanotubes did induce some fouling. FESEM images reveal some darker spots on the surface, which can be assigned to fouling (Fig. 8). The application of larger pore size membranes as support materials will be a reasonable way to avoid the effect of fouling and to gain higher fluxes.
3.6. Toxicity
The toxicity of diclofenac (25 mg L−1) treated photocatalytically and photolytically in a static setup and the toxicity of diclofenac (25 mg L−1) treated photocatalytically in a cross-flow setup was monitored (see Fig. 10). The untreated diclofenac with a concentration of 25 mg L−1 showed a high toxicity towards yeast (low cell viability). The small variance in the cell viability of the untreated diclofenac solution (25 mg L−1) of the two tests in (a) and (b) resulted from a different charge of yeast cells. The cell viability increased due to the treatment of diclofenac via photocatalysis (with the nanotubular TiO2–PES membrane and UVA-light) in the static and the cross-flow experiments (Fig. 10(a) and (b)). Treatment via photolysis (only UVA-light) in the static experiment in Fig. 10(a) also increased the cell viability, but to a lower degree compared to the photocatalysis method. After 25 min of UV exposure, the viability was almost the same for photocatalytically and photolytically treated diclofenac (16% and 15%, respectively) and also the degradation rate of diclofenac differed only by a small margin (22% by photocatalysis to 14% by photolysis of degraded diclofenac) in the static experiment. The difference between the viability/degradation value of photocatalytically and photolytically degraded diclofenac increased with proceeding UV exposure towards higher viability and degradation for the photocatalytic treatment. | ||
| Fig. 10 The cell viability of yeast cells untreated (reference) and treated with diclofenac (25 mg L−1, at minute 0) and with degraded diclofenac via photocatalysis and photolysis over time (columns) in the static setup (a) and via photocatalysis over time (columns) in the cross-flow setup (b) is displayed. The degradation of diclofenac (photocatalytically and photolytically) is also shown over time (point-line-graphs). | ||
Rizzo et al.63 as well as Achilleos et al.64 saw in the first 15 to 20 min of photocatalytic treatment an increase, and from 30 to 60 min a decrease and again at 120 min an increase in toxicity. The increase in toxicity in the first 20 min was attributed to generation of intermediate toxic products (formation of chloroderivatives),61 which were degraded after 20 min. The two studies only monitored the toxicity of diclofenac for up to 120 min of degradation, where the toxicity again increased. In this study a photocatalytic treatment up to 480 min in the static experiment and up to 21 days in the cross-flow experiment induced a further increase in cell viability instead, showing that TiO2 was able to decrease the concentration of toxic compounds on a longer time scale. It is important to completely degrade diclofenac and its intermediate products in order to gain a less toxic solution without any toxic photocatalytic/photolytic intermediates.
4. Conclusion
The nanotubular TiO2–PES membrane showed very good properties as a photocatalytically active membrane system, enabling the complete degradation of diclofenac and its toxic degradation products in a continuous way. It can be concluded that:• Cooling of the membrane at the boundary of the solution to air avoided high temperatures and the breakdown of voltage so that the generation of TiO2 nanotubes on larger membrane areas was successful.
• The TiO2 nanotubes were strongly bound to the support membrane. There was neither large breakage nor visible damage after treating the nanotubular TiO2–PES membrane with a constant stream of water horizontal to the membrane or during cross-flow filtration.
• Amorphous TiO2 nanotubes normally exhibit hydrophobic properties but become highly hydrophilic after crystallizing them with water vapor at 110 °C. This can be explained by the loss of tube spacing as the tube walls grew during water vapor crystallization.
• The degradation of diclofenac in both static and cross-flow experiments was possible. The degradation rate was higher for diclofenac solutions with a lower initial concentration. Membranes with higher pore diameters seem to be more suitable for cross-flow applications.
• The toxicity of photocatalytically or photolytically treated diclofenac towards yeast was reduced with ongoing treatment, showing a higher decrease for the photocatalytically treated diclofenac solution. It is important to fully degrade diclofenac to obtain a non-toxic product.
Acknowledgements
The authors are indebted to P. Hertel and I. Thomas for the magnetron sputtering of titanium to the membrane and TOC measurements. The financial support by the Federal State of Germany, the Free State of Saxony and the German Federal Environmental Foundation is gratefully acknowledged.Notes and references
- Y. Luo, W. Guo, H. H. Ngo, L. D. Nghiem, F. I. Hai, J. Zhang, S. Liang and X. C. Wang, Sci. Total Environ., 2014, 473–474, 619–641 CrossRef CAS PubMed.
- S. Monteiro and A. A. Boxall, Rev. Environ. Contam. Toxicol., 2010, 202, 53–154 CAS.
- Human Pharmaceuticals in the Environment, ed. B. W. Brooks and D. B. Hugget, Springer, New York, Heidelberg, Dordrecht, London, 2012 Search PubMed.
- C. G. Daughton and T. A. Ternes, Environ. Health Perspect., 1999, 107, 907–938 CrossRef CAS.
- Y. Zhang, S.-U. Geißen and C. Gal, Chemosphere, 2008, 73, 1151–1161 CrossRef CAS PubMed.
- H.-R. Buser, T. Poiger and M. D. Müller, Environ. Sci. Technol., 1998, 32, 3449–3456 CrossRef CAS.
- N. Vieno and M. Sillanpää, Environ. Int., 2014, 69, 28–39 CrossRef CAS PubMed.
- A. M. Deegan, B. Shaik, K. Nolan, K. Urell, M. Oelgmöller, J. Tobin and A. Morrissey, Int. J. Environ. Sci. Technol., 2011, 8, 649–666 CrossRef CAS.
- U. I. Gaya and A. H. Abdullah, J. Photochem. Photobiol., C, 2008, 9, 1–12 CrossRef CAS PubMed.
- M. Petrovic, J. Radjenovic and D. Barceló, Holistic Approach Environ., 2011, 1, 63–74 CAS.
- D. Gerrity and S. Snyder, in Human Pharmaceuticals in the Environment – Current and Future Perspectives, ed. B. W. Brooks and D. B. Huggett, Springer, New York, 2012, p. 225 Search PubMed.
- Y. Gao, M. Hu and B. Mi, J. Membr. Sci., 2014, 455, 349–356 CrossRef CAS PubMed.
- R. Molinari, F. Pirillo, V. Loddo and L. Palmisano, Catal. Today, 2006, 118, 205–213 CrossRef CAS PubMed.
- R. Molinari, L. Palmisano, E. Drioli and M. Schiavello, J. Membr. Sci., 2002, 206, 399–415 CrossRef CAS.
- S. Mozia, Sep. Purif. Technol., 2010, 73, 71–91 CrossRef CAS PubMed.
- H. Jiang, G. Zhang, T. Huang, J. Chen, Q. Wang and Q. Meng, Chem. Eng. J., 2010, 156, 571–577 CrossRef CAS PubMed.
- V. C. Sarasidis, K. V. Plakas, S. I. Patsios and A. J. Karabelas, Chem. Eng. J., 2014, 239, 299–311 CrossRef CAS PubMed.
- N. K. O. Cruz, G. U. Semblante, D. B. Senoro, S.-J. You and S.-C. Lu, J. Taiwan Inst. Chem. Eng., 2014, 45, 192–201 CrossRef CAS PubMed.
- K. Nakata and A. Fujishima, J. Photochem. Photobiol., C, 2012, 13, 169–189 CrossRef CAS PubMed.
- K. Zhu, N. R. Neale, A. Miedaner and A. J. Frank, Nano Lett., 2006, 7, 69–74 CrossRef PubMed.
- G. K. Mor, K. Shankar, M. Paulose, O. K. Varghese and C. A. Grimes, Nano Lett., 2004, 5, 191–195 CrossRef PubMed.
- W. Krengvirat, S. Sreekantan, A.-F. Mohd Noor, N. Negishi, G. Kawamura, H. Muto and A. Matsuda, Mater. Chem. Phys., 2013, 137, 991–998 CrossRef CAS PubMed.
- R. A. Damodar, S.-J. You and H.-H. Chou, J. Hazard. Mater., 2009, 172, 1321–1328 CrossRef CAS PubMed.
- V. C. Sarasidis, S. I. Patsios and A. J. Karabelas, Sep. Purif. Technol., 2011, 80, 73–80 CrossRef CAS PubMed.
- H. Song, J. Shao, Y. He, B. Liu and X. Zhong, J. Membr. Sci., 2012, 405–406, 48–56 CrossRef CAS PubMed.
- K. Fischer, R. Gläser and A. Schulze, Appl. Catal., B, 2014, 160–161, 456–464 CrossRef CAS PubMed.
- L. Rumlova and J. Dolezalova, Environ. Toxicol. Pharmacol., 2012, 33, 459–464 CrossRef CAS PubMed.
- H. P. Koch, M. Hofeneder and B. Bohne, Methods Find. Exp. Clin. Pharmacol., 1993, 15, 141 CAS.
- C.-Q. Jing, Y.-T. Liu, X.-Y. Shen, L. Li, Q.-Y. Gao and Y. Yu, Toxin Rev., 2012, 31, 27–31 CrossRef CAS.
- K. Estève, C. Poupot, P. Dabert, M. Mietton-Peuchot and V. Milisic, J. Ind. Microbiol. Biotechnol., 2009, 36, 1529–1534 CrossRef PubMed.
- A. Kungolos, I. Aoyama and S. Muramoto, Ecotoxicol. Environ. Saf., 1999, 43, 149–155 CrossRef CAS PubMed.
- A. Schulze, B. Marquardt, S. Kaczmarek, R. Schubert, A. Prager and M. R. Buchmeiser, Macromol. Rapid Commun., 2010, 31, 467–472 CrossRef CAS PubMed.
- D. Hong, G. Lee, N. C. Jung and M. Jeon, Biol. Proced. Online, 2013, 15, 13 CrossRef PubMed.
- Y. Liao, J. Brame, W. Que, Z. Xiu, H. Xie, Q. Li, M. Fabian and P. J. Alvarez, J. Hazard. Mater., 2013, 260, 434–441 CrossRef CAS PubMed.
- J. M. Macák, H. Tsuchiya and P. Schmuki, Angew. Chem., Int. Ed., 2005, 44, 2100–2102 CrossRef PubMed.
- S. P. Albu, A. Ghicov, J. M. Macák and P. Schmuki, Phys. Status Solidi RRL, 2007, 1, R65–R67 CrossRef CAS PubMed.
- Y. Tang, J. Tao, Z. Dong, J. T. Oh and Z. Chen, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2011, 2, 045002 CrossRef.
- M. Paulose, H. E. Prakasam, O. K. Varghese, L. Peng, K. C. Popat, G. K. Mor, T. A. Desai and C. A. Grimes, J. Phys. Chem. C, 2007, 111, 14992–14997 CAS.
- C. A. Grimes, J. Mater. Chem., 2007, 17, 1451–1457 RSC.
- Y. Li, Q. Ma, J. Han, L. Ji, J. Wang, J. Chen and Y. Wang, Appl. Surf. Sci., 2014, 297, 103–108 CrossRef CAS PubMed.
- P. Acevedo-Peña, L. Lartundo-Rojas and I. González, J. Solid State Electrochem., 2013, 1–9 Search PubMed.
- D. K. Platt, Engineering and High Performance Plastics – Market Report, Rapra Technology Limited, 2003 Search PubMed.
- G. F. Ortiz, I. Hanzu, P. Knauth, P. Lavela, J. L. Tirado and T. Djenizian, Electrochim. Acta, 2009, 54, 4262–4268 CrossRef CAS PubMed.
- Y.-x. Tang, J. Tao, Y.-y. Zhang, T. Wu, H.-j. Tao and Y.-r. Zhu, Trans. Nonferrous Met. Soc. China, 2009, 19, 192–198 CrossRef CAS.
- O. K. Varghese, M. Paulose and C. A. Grimes, Nat. Nanotechnol., 2009, 4, 592–597 CrossRef CAS PubMed.
- H. Nanjo, F. M. B. Hassan, S. Venkatachalam, N. Teshima, K. Kawasaki, T. Aizawa, T. Aida and T. Ebina, J. Power Sources, 2010, 195, 5902–5908 CrossRef CAS PubMed.
- V. Galstyan, A. Vomiero, E. Comini, G. Faglia and G. Sberveglieri, RSC Adv., 2011, 1, 1038–1044 RSC.
- V. Galstyan, A. Vomiero, I. Concina, A. Braga, M. Brisotto, E. Bontempi, G. Faglia and G. Sberveglieri, Small, 2011, 7, 2437–2442 CrossRef CAS PubMed.
- S. L. Lim, Y. Liu, J. Li, E.-T. Kang and C. K. Ong, Appl. Surf. Sci., 2011, 257, 6612–6617 CrossRef CAS PubMed.
- A. Vomiero, V. Galstyan, A. Braga, I. Concina, M. Brisotto, E. Bontempi and G. Sberveglieri, Energy Environ. Sci., 2011, 4, 3408–3413 CAS.
- J. Weickert, C. Palumbiny, M. Nedelcu, T. Bein and L. Schmidt-Mende, Chem. Mater., 2011, 23, 155–162 CrossRef CAS.
- J. Ni, C. J. Frandsen, K. Noh, G. W. Johnston, G. He, T. Tang and S. Jin, Mater. Sci. Eng., C, 2013, 33, 1460–1466 CrossRef CAS PubMed.
- N.-S. Jang, M. S. Kim, S.-H. Kim, S.-K. Lee and J.-M. Kim, Sens. Actuators, B, 2014, 199, 361–368 CrossRef CAS PubMed.
- J. Macák, H. Tsuchiya, S. Berger, S. Bauer, S. Fujimoto and P. Schmuki, Chem. Phys. Lett., 2006, 428, 421–425 CrossRef PubMed.
- C. E. Scott, Poly(ethylene terephthalate), http://www.polymerprocessing.com/polymers/PET.html, accessed 24 September 2014.
- P. S. G. Krishnan and S. T. Kulkarni, in Polyester and polyamides, ed. B. L. Deopura, R. Alagirusamy, M. Joshi and B. Gupta, Woodhead Publishing Limited in association with The Textile Institute, Raton, USA, 2008, vol. 1, p. 27 Search PubMed.
- D. H. Shin, T. Shokuhfar, C. K. Choi, S.-H. Lee and C. Friedrich, Nanotechnology, 2011, 22, 315704 CrossRef PubMed.
- A. Hamlekhan, A. Butt, S. Patel, D. Royhman, C. Takoudis, C. Sukotjo, J. Yuan, G. Jursich, M. T. Mathew, W. Hendrickson, A. Virdi and T. Shokuhfar, PLoS One, 2014, 9, 1–10 Search PubMed.
- S. Yoriya, W. Kittimeteeworakul and N. Punprasert, J. Chem. Chem. Eng., 2012, 6, 686 CAS.
- J. Zhao, X. Wang, T. Sun and L. Li, J. Alloys Compd., 2007, 434–435, 792–795 CrossRef CAS PubMed.
- P. Calza, V. A. Sakkas, C. Medana, C. Baiocchi, A. Dimou, E. Pelizzetti and T. Albanis, Appl. Catal., B, 2006, 67, 197–205 CrossRef CAS PubMed.
- F. Méndez-Arriaga, S. Esplugas and J. Gimenez, Water Res., 2008, 42, 585–594 CrossRef PubMed.
- L. Rizzo, S. Meric, D. Kassinos, M. Guida, F. Russo and V. Belgiorno, Water Res., 2009, 43, 979–988 CrossRef CAS PubMed.
- A. Achilleos, E. Hapeshi, N. P. Xekoukoulotakis, D. Mantzavinos and D. Fatta-Kassinos, Chem. Eng. J., 2010, 161, 53–59 CrossRef CAS PubMed.
- I. Epold, N. Dulova and M. Trapido, J. Environ. Eng. Ecol. Sci., 2012, 1 Search PubMed.
- M. E. Palomo, M. P. Ballesteros and P. Frutos, J. Pharm. Biomed. Anal., 1999, 21, 83–94 CrossRef CAS.
- M. Kincl, M. Meleh, M. Veber and F. Vrečerd, Acta Chim. Slov., 2004, 51, 409–425 CAS.
- P. Bartels and W. von Tümpling Jr, Sci. Total Environ., 2007, 374, 143–155 CrossRef CAS PubMed.
- M. Luo, Q. Wen, J. Liu, H. Liu and Z. Jia, Chin. J. Chem. Eng., 2011, 19, 45–51 CrossRef CAS.
- C. Zhao, J. Xue, F. Ran and S. Sun, Prog. Mater. Sci., 2013, 58, 76–150 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |