
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrothermal synthesis of single crystal CoAs2O4 and NiAs2O4 compounds and their magnetic properties†
Tamara
Đorđević
*a,
Astrid
Wittwer
a,
Zvonko
Jagličić
bc and
Igor
Djerdj
d
aInstitut für Mineralogie und Kristallographie, Universität Wien, Althanstraße 14, A-1090 Wien, Austria. E-mail: tamara.djordjevic@univie.ac.at
bInstitute of Mathematics, Physics and Mechanics, Jadranska 19, 1000, Ljubljana, Slovenia
cFaculty of Civil and Geodetic Engineering, University of Ljubljana, Jamova 2, 1000 Ljubljana, Slovenia
dRuđer Bošković Institute, Bijenička 54, 10000 Zagreb, Croatia
First published on 5th February 2015
Abstract
The crystal structures of the hydrothermally synthesised trippkeite-related materials, CoAs2O4 and NiAs2O4 were investigated by means of single-crystal X-ray diffraction, Raman and infrared spectroscopy. The obtained compounds crystallise in the tetragonal crystal system (P42/mbc), with unit cell parameters at 293 K of a = 8.34530(10)/8.2277(12) Å, c = 5.62010(10)/5.6120(11) Å, V = 391.406(12)/379.90(13) Å3, Z = 4, for CoAs2O4 and NiAs2O4 respectively. Magnetic measurements show that the resulting single crystal of NiAs2O4 exhibits an antiferromagnetic transition at TN = 53 K in a high magnetic field of 10 kOe, as already reported in the literature. The single crystal of CoAs2O4 reveals an interplay between ferromagnetic and a canted antiferromagnetic interactions resulting in a canted antiferromagnetic state which occurs at 105 K – the highest critical temperature among all similar structures.
1. Introduction
In order to recognise the role of arsenic in the environment, one has to investigate structures and stabilities of naturally occurring arsenic compounds. Besides, a study of mineral-related synthetic phases should be useful, because they can appear as a consequence of human activities. The susceptibility of As(III) to oxidation to As(V) in oxide environments affords high thermal stability only to ternary X–As(V)–O oxides (X = Mg and divalent 3d transition metal) with arsenic in its higher oxidation state. However, under strict synthetic conditions, X–As(III)–O oxides can be formed. For that reason, anhydrous arsenates of cobalt and nickel are well-established but arsenites of cobalt and nickel are less known. Besides Co2As2O5,1 CoAs2O4 represents the second compound in the Co(II)–As(III)–O system and the NiAs2O4 is the first structurally determined compound in the Ni(II)–As(III)–O system.CoAs2O4 and NiAs2O4 are isostructural to the M2+X23+O4 materials and minerals,2–9 with the exception of ZnAs2O4.3 These compounds crystallise tetragonal, adopting space group P42/mbc, and contain chains of edge-linked MO6 octahedra running along [001]; the chains are connected via trigonal pyramidal XO3 units.
All MSb2O4 phases, with the exception of MgSb2O4,4 have been shown to display antiferromagnetic ordering with Néel temperatures in the range 40–60 K and a transition of the magnetic modal ordering from a predominant A mode to a C mode (vide infra) on crossing the first row transition metals.5,7,8,10 The edge sharing nature of the octahedra and the superexchange interactions between the M2+ transition metals in the chains and between the chains are responsible for the magnetic properties of these group compounds. However, no crystallographic and magnetic structures have been reported for the M2+As2O4 (M2+ = Co, Ni) compounds. Molecular susceptibility of NiAs2O4 has been measured and the values of the Néel temperature and the asymptotic Curie temperature are given.11
CoAs2O4 and NiAs2O4 were synthesised during an on-going research on natural and synthetic arsenic oxo-salts, with a focus on their structural and spectroscopic classification. The present article reports the hydrothermal synthesis of two new arsenites, CoAs2O4 and NiAs2O4. The results of the determination of their crystal structures based on single-crystal X-ray diffraction data are given and the relationship to the known M2+X2O4 compounds is discussed. To obtain further information on anion groups, Raman and infrared spectra were acquired. Due to the presence of transition metal M2+ cations, non-diamagnetic ground state of as grown crystals is expected and investigated using SQUID measurements. Continuous investigations on the crystal chemistry of the arsenic oxo-salts are performed because arsenic is at the top of the priority of the most hazardous substances, but less is known about its crystal structures.
2. Experimental
2.1 Materials
The materials used were: KCl (Loba Chemie, 13508), Co(OH)2 (Sigma-Aldrich, 342440, tech. 95%), Ni(NO3)2 (Sigma-Aldrich 244074-500G), As2O3 (Alfa Aesar 33289, ACS 99.95–100.05%).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1. The initial pH value of the mixture was 6. The stainless-steel autoclave was then closed and the crystallisation was carried out by placing the autoclave in oven under air atmosphere and heating the mixture under autogenous pressure from room temperature. A heating regime with three steps was chosen: the autoclaves were heated from 20 °C to 200 °C (four hours), held at 200 °C for 199 h, and finally cooled to room temperature within 98 h. The pH value of supernatant solution was 6. The obtained products were washed with distilled water, filtered and dried in the air at room temperature. CoAs2O4 crystallised as pink transparent elongated prisms (yield 60%) (Fig. 1a) together with transparent crystals of KAs4O6Cl12 (yield 40%). The maximal length of crystals was about 2 mm.
:1:1. The initial pH value of the mixture was 6. The stainless-steel autoclave was then closed and the crystallisation was carried out by placing the autoclave in oven under air atmosphere and heating the mixture under autogenous pressure from room temperature. A heating regime with three steps was chosen: the autoclaves were heated from 20 °C to 200 °C (four hours), held at 200 °C for 199 h, and finally cooled to room temperature within 98 h. The pH value of supernatant solution was 6. The obtained products were washed with distilled water, filtered and dried in the air at room temperature. CoAs2O4 crystallised as pink transparent elongated prisms (yield 60%) (Fig. 1a) together with transparent crystals of KAs4O6Cl12 (yield 40%). The maximal length of crystals was about 2 mm.
 | ||
| Fig. 1 Single crystals of (a) CoAs2O4 and (b) NiAs2O4. | ||
:1. The initial pH value of the mixture was kept at around 2.5 in order to avoid the oxidation of As3+ to As5+. A heating regime with three steps was chosen: the autoclaves were heated from 20 °C to 220 °C (four hours), held at 220 °C for 56 h, and slowly cooled to room temperature within 320 h. The pH value of supernatant solution was 4. The products obtained were washed with distilled water, filtered and dried in the air at room temperature. NiAs2O4 crystallised as green transparent elongated prisms (yield ca. 95%) (Fig. 1b). The maximal length of crystals was about 1.7 mm.
2.2 Single-crystal X-ray diffraction measurements
Several single crystals of the two title compounds were studied on a Bruker AXS Kappa APEX II CCD diffractometer, equipped with a monocapillary optics collimator and graphite-monochromatised MoKα radiation (λ = 0.71073 Å). Single-crystal X-ray diffraction data were collected at room temperature, integrated and corrected for Lorentz and polarization factors and absorption correction by evaluation of partial multiscans (see Table 1 for details). The intensity data were processed with the Bruker-Nonius programme suite SAINT-Plus13 and corrected for Lorentz, polarization, background and absorption effects. Their crystal structures were refined with SHELXL-97 (ref. 14) and WinGX15 starting from the atomic coordinates given for isotypic CuAs2O4.2| Crystal data | ||
|---|---|---|
| a w = 1/[σ2(Fo2) + (0197P)2 + 0.1338P] for CoAs2O4, w = 1/[σ2(Fo2) + (0.0129P)2 + 0.2839P] for NiAs2O4. | ||
| Chemical formula | CoAs2O4 | NiAs2O4 |
| Temperature (K) | 293(2) | 293(2) |
| Crystal form, colour | Prismatic, pink | Prismatic, green |
| Formula weight, Mr (g mol−1) | 272.77 | 272.55 |
| System, space group (no.) | Tetragonal, P42/mbc (135) | |
| a (Å) | 8.34530(10) | 8.2277(12) |
| c (Å) | 5.62010(10) | 5.6120(11) |
| V (Å3) | 391.406(12) | 379.90(13) |
| Z | 4 | 4 |
| F(000) | 500 | 504 |
| Calculated density, Dx (g cm−3) | 4.629 | 4.765 |
| Absorption coefficient, μ (mm−1) | 21.032 | 22.258 |
| Transmission factors, Tmin/Tmax | 0.124/0.678 | 0.135/0.664 |
| Crystal size (mm) | 0.02 × 0.02 × 0.17 | 0.02 × 0.02 × 0.15 |
| Reflections collected/unique | 11599/838 |
7925/595 |
| Observed reflections [I > 2 σ(I)] | 719 | 532 |
| R int | 0.0418 | 0.0382 |
| Range for data collection, θ (°) | 3.452–44.387 | 3.502–39.118 |
| Range of Miller indices | −15 ≤ h ≤ 16; −16 ≤ k ≤ 16; −10 ≤ l ≤ 9 | −14 ≤ h ≤ 14; −8 ≤ k ≤ 12; −9 ≤ l ≤ 9 |
| Extinction coefficient, ka | 0.0159(8) | 0.0056(5) |
| Refined parameters | 22 | 22 |
| R-indices [I > 2 σ(I)]a | R 1 = 0.0229 | R 1 = 0.0161 |
| wR2 = 0.0440 | wR2 = 0.0363 | |
| R-indices (all data)a | R 1 = 0.0306 | R 1 = 0.0371 |
| wR2 = 0.0455 | wR2 = 0.059 | |
| Goodness-of-fit, S | 1.079 | 1.09 |
| (Δρ)max, (Δρ)min (e Å−3) | 1.088, −1.022 | 0.490, −0.656 |
Relevant information on crystal data, data collection, and refinements are compiled in Table 1. For final positional and displacement parameters see CIF files.† Selected bond lengths and angles for both arsenites are presented in Table 2.
| CoAs2O4 | NiAs2O4 | ||
|---|---|---|---|
| a Symmetry codes: (i) y − 1/2, x + 1/2, −z + 1/2; (ii) −x, −y + 1, −z; (iii) −y + 1/2, −x + 1/2, z + 1/2; (iv) −x, −y + 1, z; (v) −x + 1/2, y − 1/2, −z; (vi) x, y, −z. | |||
| Co–O2 | 2.0305(7) | Ni–O2 | 2.0079(9) |
| –O2i | 2.0305(7) | –O2i | 2.0079(9) |
| –O2ii | 2.0305(7) | –O2ii | 2.0079(9) |
| –O2iii | 2.0305(7) | –O2iii | 2.0079(9) |
| –O1 | 2.1775(11) | –O1 | 2.1222(13) |
| –O1iv | 2.1775(11) | –O1iv | 2.1222(13) |
| 〈Co–O〉 | 2.0795 | 〈Ni–O〉 | 2.046 |
| As–O2v | 1.7292(11) | As–O2v | 1.7275(12) |
| –O1vi | 1.8473(5) | –O1vi | 1.8501(6) |
| –O1 | 1.8473(5) | –O1 | 1.8501(6) |
| 〈As–O〉 | 1.8079 | 〈As–O〉 | 1.8092 |
| Co–Co | 2.81005(5) | Ni–Ni | 2.8060(5) |
| ∠O1b–As–O2t | 96.76(3) × 2 | ∠O1b–As–O2t | 96.64(4) × 2 |
| ∠O1b–As–O1b | 99.03(4) | ∠O1b–As–O1b | 98.63(4) |
| ∠As–O1b–As | 124.09(6) | ∠As–O1b–As | 124.13(7) |
2.3 Vibrational spectroscopy measurements
The single-crystal Raman spectra and Raman spectra of the bulk material of CoAs2O4 and NiAs2O4 were collected using a Horiba LabRam HR Evolution system equipped with a Si-based, Peltier-cooled CCD detector in the spectral range from 4000 to 60 cm−1. The 633 nm excitation line of a He–Ne laser was focused with a 100× objective on the randomly oriented single crystal. The sample spectra were acquired with a nominal exposure time of 5 s and 10 s, for Co- and Ni–arsenite, respectively (confocal mode, Olympus 1800 lines per mm, 1.5 μm lateral resolution, approximately 3 μm depth resolution).Fourier transform infrared (FTIR) absorption spectra of the title compounds were recorded using a Bruker Tensor 27 FTIR spectrophotometer, equipped with mid-IR Globar light source and KBr beam splitter, attached to a Hyperion 2000 FTIR microscope with liquid nitrogen-cooled mid-IR, broad-band MCT detector. A total of 128 scans were accumulated between 4000 and 370 cm−1 using the samples prepared as KBr pellets (KBr:MAs2O4 = 200:1).
2.4 Magnetic measurements
The magnetisation was measured with a QUANTUM DESIGN MPMS-XL-5 SQUID magnetometer. Zero-field-cooled (ZFC) and field-cooled (FC) runs were performed between room temperature and 2 K in a static magnetic field of 10 kOe, 1 kOe and 100 Oe. Isothermal magnetization curves were measured at several temperatures below and above the critical temperature.3. Results and discussion
3.1 Crystal structures
In the crystal structures of MAs2O4, the chains of edge-sharing MO6 octahedra run parallel to the [001] direction, where the individual octahedra are orientated such that the apical, M–O1 bonds lie perpendicular to [001], and are directed towards the adjacent chain, which are further interconnected with the chains of corner sharing (AsO3)3− groups (Fig. 2 and 3). | ||
| Fig. 2 Part of the crystal structure of the tetragonal M2+X23+O4 compounds showing the connection of the MO6 octahedral chains via trigonal ψ-(AsO3)3− pyramids. | ||
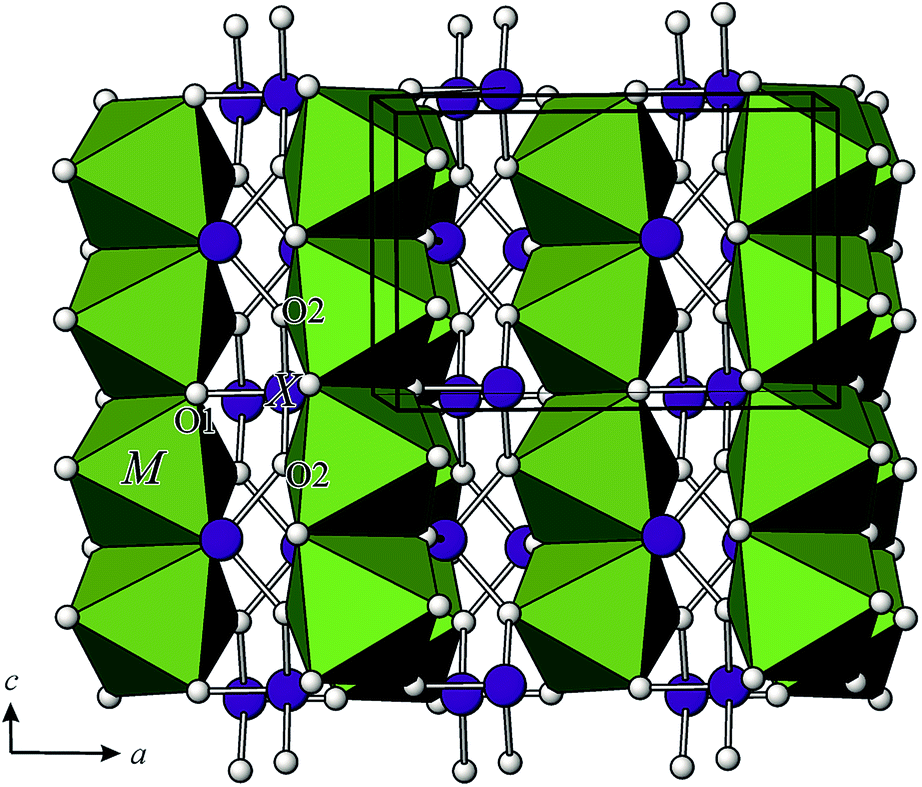 | ||
| Fig. 3 Perspective view of the crystal structure of the tetragonal M2+X23+O4 compounds. | ||
The average Co–O and Ni–O bond lengths are 2.0795 and 2.046 Å. According to the formula Δoct = 1/6 Σ[(di − dm)/dm]2 the bond-length distortions for the Co and Ni atoms amount to 1.11 × 10−3 and 6.92 × 10−4, respectively and indicate large distortions.16,17 These results compare well with the values compiled by Wildner18 for CoO6 octahedra in accurately determined crystal structures, who found 672 Co–O bond lengths between 1.959 and 2.517 Å. The average 〈Co–O〉 bond lengths for 112 polyhedra are in the range of 2.054 to 2.182 Å; the overall mean value is 2.1115 Å.
The MO6 octahedra are edge-linked to chains parallel to the 4-fold axis. The shared edges O2–O2i (i = −x, −y + 1, −z) have lengths of 2.932(2) and 2.873(2) Å, for Co and NiAs2O4, respectively. Due to this connection, the angular distortion is large: σoct2 = 1/11 Σ(∠i − 90)2 is 20.59 and 17.49 for the two CoO6 and NiO6 octahedra, respectively.15,16 The inter-transition metal separation distances, Co–Co and Ni–Ni along [001] are equal to the c/2 and amount 2.81005(5) and 2.8060(5) Å.
Arsenic is one-sided coordinated to three oxygen atoms and its coordination figure is represented by ψ-(AsO3)3− pyramids (ψ is a stereoactive lone pair of electrons), where the As atoms lies at the vertex and the oxygen atoms are at the basis of the pyramid. ψ-(AsO3)3− pyramids share corners thus forming chains also parallel to the 4-fold axis. Oxygen atoms of the pyramid basis lie parallel to the vertex and the oxygen atoms are at the basis of the pyramid. ψ-(AsO3)3− pyramids share corners thus forming chains also parallel to the 4-fold axis. Oxygen atoms of the pyramid basis lie parallel to the (110) plane, and the As-atoms alternate left and right from that plane (Fig. 3). The As–O bond lengths within the single chain are significantly longer (1.8473(5) and 1.8501(6) Å, respectively) than the third As–O bond length (1.7292(11) and 1.7275(12) Å, respectively). As–O1b–As angles within a single chain are 124.09(6)° and 124.13(7)° for CoAs2O4 and NiAs2O4, respectively and compare well with the As–Ob–As angles in other ‘chain’ arsenites [127.3(3)° in CuAs2O4,2 from 123.3(5) to 125.1(3)° in XAsO2,19 (X = Na, K, Rb), 125.1(14)° in Cs3As5O9 (ref. 18) and 121.3(5) and 122.3(5) in leitite, ZnAs2O4.3
Bond valence sum calculations20 based on the room temperature data show the models to be compatible with the bonding requirements of Co2+ (2.10 v.u.), Ni2+ (2.08 v.u.) and As3+ (2.88/2.88 v.u.) O1 (2.24/2.26 v.u.), O2 (1.98/1.94 v.u.) for CoAs2O4 and NiAs2O4, respectively.
3.2 Vibrational spectra analysis
Spectroscopic data on arsenites that have been previously published are so far rather incomplete and not in good agreement with each other. However, the spectra assignments of CoAs2O4 and NiAs2O4 may be based on the single-crystal Raman study of synthetic trippkeite, CuAs2O4 (ref. 21) and AAsO2(ref. 19) (A = Na, K, and Rb). In the AAsO2 compounds, where AsO3 units are also interconnected to the chains (each unit possessing one terminal O and two bridging O atoms), the bands above 800 cm−1 are observed in each spectrum. These bands were assigned to the vibration of terminal oxygen atoms. The bands at 810 and 780 cm−1 in synthetic trippkeite are assigned to stretches of terminal O atoms and stretches of the bridging O atoms are assigned to bands at 657 and 496 cm−1. Therefore the distinct frequency ranges in CoAs2O4 and NiAs2O4 may be assigned as follows:The Raman and infrared (IR) spectra of CoAs2O4 and NiAs2O4 are presented at the Fig. 4–6.
 | ||
| Fig. 4 The single-crystal Raman spectra of CoAs2O4 and NiAs2O4 presented in two different orientations. p = elongation of the crystal is parallel to the laser beam, and n = elongation of the crystal is normal to the laser beam. | ||
 | ||
| Fig. 5 Raman spectra of the bulk CoAs2O4 and NiAs2O4 crystals. | ||
 | ||
| Fig. 6 Infrared spectra of CoAs2O4 and NiAs2O4. | ||
The Raman spectra of both title compounds were obtained aligning the laser beam parallel and normal to the longest axis of the single-crystal. The strong bands at 776 and 774 cm−1 (778 cm−1 in IR-spectrum and 783 cm −1 in the Raman spectrum of the bulk) in NiA2O4 and 787 and 780 cm−1 (767 cm−1 in IR-spectrum and 769 cm −1 in the Raman spectrum of the bulk) in CoAs2O4 in parallel (p) and normal (n) orientation to the laser beam, respectively may be assigned to the symmetric As–Oterminal stretches, and the weak bands between 700 and 450 cm−1 in both orientations and the bulk Raman spectra are assigned to the As–Obridging stretches (strong bands at 533 and 494 cm−1 and 540, 516, 486 cm−1 in IR-spectrum of NiAs2O4 and CoAs2O4, respectively). In the p orientation of the Raman spectra, bands of very low intensity were observed at 960 and 958 cm−1 (around 960 cm−1 in IR spectra) as well as a shoulder to the bands around 780 cm−1 at 831 and 822 cm−1 in NiAs2O4 and CoAs2O4, respectively. These bands are attributed also to the symmetric As–Oterminal stretches. The very weak bands being seen only in p orientation at 747 and at the bulk spectrum (strong band at 753 cm−1) and 646 cm−1 in NiAs2O4 and 638 cm−1 (strong band at 739 cm−1) in CoAs2O4, respectively, may be attributed to the antisymmetric stretches. It is suggested that the (AsO3)3− group is the only tetrahedral oxyanion of the main group elements in which νs > νas.22 The same is suggested for (As2O4)2− group by Bencivenni and Gingerich.23 These authors noted that it was unusual for the vibrational spectroscopy of oxy-anions. Further strong bands in the spectra of both arsenites are around 350 cm−1 (357 and 352 cm−1 in NiAs2O4 and 354 and 347 cm−1 in CoAs2O4) and are assigned to the ν4 As2O42− bending modes. Below 300 cm−1 appear the various lattice modes of the compounds. The structure of Co- and NiAs2O4 consists of arrays of As–O–As–O–As chains. Two types of components are predicted based upon OAsO and AsOAs units. The bands between 300 and 200 cm−1 in the both compounds are assigned to the AsOAs linkages.24
3.3 Magnetic properties
The magnetic properties of the NiAs2O4 and CoAs2O4 single crystals were investigated, since the presence of transition metals in chemical composition with unpaired d electrons indicates the non-diamagnetic ground state. All single crystals used for the magnetic measurements were studied by single-crystal X-ray diffraction techniques. The measurements showed the same primitive tetragonal unit cell, without additional non-indexed reflections. All single crystals have been probed in the constant magnetic field oriented with their c-axis parallel to the magnetic field (H ‖ c), while the crystals of CoAs2O4 have been measured also in H ⊥ c orientation. The temperature dependence of the inverse molar magnetic susceptibility (measured at 10 kOe and displayed in Fig. 7) has a typical paramagnetic shape between room temperature and approximately 140 K, and can be described in the high temperature range (140–300 K) with the Curie–Weiss law: χ(T) = χ0 + C/(T − θ), where χ0 is the temperature-independent part of χ, i.e. diamagnetic contribution, C is the Curie constant, and θ is the Curie–Weiss temperature. From the slope of χ−1vs. T graph we obtain peff = 3.3 μB and Curie–Weiss temperature θ of approximately −40 K for NiAs2O4 as reported in Witteveen,11 while peff = 5.6 μB and θ ≈ 75 K for CoAs2O4 sample. The paramagnetic effective moments peff calculated from the Curie constant are in a close agreement with the literature data for Ni2+ cation (3.2 μB) and high spin Co2+ cation with large orbital contribution (6.5 μB).25 Below 140 K the susceptibility of CoAs2O4 sample starts to deviate from paramagnetic (linear χ−1vs. T) regime, while for NiAs2O4 the same occurs below 70 K. We also performed an equivalent analysis of inverse molar magnetic susceptibility measured under external field of 1 kOe, and obtained results are practically identical as for previously described measurement (Fig. S1 – ESI†). | ||
| Fig. 7 Temperature dependence of the inverse magnetic susceptibility for the studied NiAs2O4 and CoAs2O4 single crystals measured along crystal c-axis in a DC field H = 10 kOe. The solid (black) lines are the best fit to the Curie–Weiss law at high temperatures. | ||
Magnetic susceptibility measurements of NiAs2O4 under ZFC and FC conditions (Fig. 8a) show some unexpected results. At TN = 53 K in high magnetic field of 10 kOe, the susceptibility shows behavior consistent with a transition to the antiferromagnetic ground state without splitting between ZFC and FC curves as already reported.24 When the applied DC field is ten times lower, at the same temperature ZFC and FC curves start to diverge. Such divergence is highly enhanced when applied field was 100 Oe only. This first observed strong dependence of the transition at 55 K on magnetic field might be a consequence of two different superexchange interactions between Ni2+ ions: a positive and weaker one intrachain J1 between magnetic moments in the chain, and a negative and stronger interchain interaction J1 as proposed by Witteveen.11 The unexpected behaviour of ZFC-FC curves is detected at the temperature around 20 K, where the transition to the ferromagnetic state has been clearly observed. Finally, below 15 K ZFC-FC curves show a strong divergence.
 | ||
| Fig. 8 (a) ZFC and FC molar magnetization versus temperature curves for NiAs2O4 measured in the various magnetic fields aligned along crystal c-axis. (b) Magnetization versus applied field aligned along crystal c axis for NiAs2O4 at 2, 15, 50 and 70 K. | ||
This second magnetic transition influences also the magnetization curves as only at 2 K where a small hysteresis with coercivity Hc = 1.6 kOe and remanent magnetization Mr = 7.5 × 10−3μB/Ni atom can be observed in M–H graph displayed in Fig. 8b. M(H) curve measured at 50 K and 70 K is rather linear.
The phase transition from paramagnetic to magnetic ordered phase in CoAs2O4 occurs at higher temperature as in NiAs2O4. The positive Curie–Weiss temperature ϑ = 75 K obtained from χ−1vs. T plot suggests a ferromagnetic interaction between cobalt ions. Indeed, with decreasing temperature, the susceptibility measured in H = 100 Oe suddenly sharply increases at Tc = 105.5 K. The critical temperature Tc was defined from the fit M ∝ (1 − T/Tc)β as shown in inset in Fig. 9a. The obtained critical exponent β = 0.34 agrees with the theoretically calculated value for 3-D Ising system.26 The susceptibility at Tc and below behaves quite differently when measured in 1 kOe or 10 kOe instead of 100 Oe. It decreases below Tc as it is characteristic for antiferromagnetic transitions. The magnetisation curves M vs. H measured at several temperatures (Fig. 9b) also show this duality – ferromagnetic and antiferromagnetic behaviour. M(H) obtained at 2 K and 10 K exhibits an “S”-shaped curve for small magnetic fields that saturates at ≈0.01 μB/(Co ion) in a field of approx. 5 kOe. In larger magnetic fields magnetisation increases linearly with the fields as expected for antiferromagnetically coupled magnetic moments.
 | ||
| Fig. 9 (a) ZFC and FC molar magnetization versus temperature curves for CoAs2O4 measured in the various magnetic fields aligned along crystal c-axis. Full line in inset is a fit M ∝ (1 − T/Tc)β. (b) Magnetization versus applied field aligned along crystal c axis for CoAs2O4 at 2 K, 10 K, and 120 K (inset). | ||
The crystal structure of CoAs2O4 compound and the distribution of magnetic Co2+ ions in chains are similar to already reported NiAs2O4 (ref. 22) (TN = 53.5 K), NiSb2O4 (ref. 22) (TN = 47 K), MnSb2O4 (ref. 5) (TN = 55 K), and CoSb2O4 (ref. 8) (TN = 79 K). In the case of CoAs2O4 the magnetic ions Co2+ possess the largest magnetic moment among above mentioned compounds with the exception of CoSb2O4 where it is roughly the same. This may be the reason why the transition to magnetically ordered phase is shifted to a higher temperature for CoAs2O4. Practically the same critical exponent as we obtained for CoAs2O4 (β = 0.34) was measured in MnSb2O4 (ref. 5) (β = 0.36) too. Having these similar structural and detected magnetic properties in mind, we propose the same – a canted antiferromagnetic structure of CoAs2O4 as described for MnSb2O4.5 Such a structure is in agreement with a measured ferromagnetic response in a small magnetic field and prevailing antiferromagnetism when measured in magnetic field of 1 kOe or larger. In order to confirm the proposed magnetic structure neutron diffraction data are needed, for which there is not enough material at the moment.
Below 10 K a similar increase of the susceptibility in CoAs2O4 can be observed as already described for NiAs2O4 below 20 K. At the moment we have no reliable explanation for these two increases of susceptibilities. Similar anomaly in the ZFC/FC susceptibility below about 20 K has been already detected for NiO nanoparticles and bulk materials.27–29
The anomaly was contributed to surface spin magnetism due to Ni2+ magnetic moments that are not coordinated in the same way as expected for the titled compound. We tentatively ascribe the measured anomalies at 20 K and 10 K in NiAs2O4 and CoAs2O4, respectively, to the surface spins. In order to test this hypothesis much larger single crystals as we used in our research are needed.
The single-crystalline nature of investigated compounds points a further magnetic research into the possible magnetic anisotropy detection. In high magnetic field (1 T) the parallel and perpendicular susceptibilities, as shown in the inset of Fig. 10, are as expected for typical two-dimensional (layered) antiferromagnetic system as already described for BaNi2(PO4)2 and Rb2Co0.7Mg0.3F4.26 While in small magnetic field of 100 Oe susceptibilities in both orientations of the sample increases below TN. The increase is even larger for perpendicular orientation, in agreement with our hypothesis of canted magnetic moments from c-direction.
 | ||
| Fig. 10 FC molar magnetization versus temperature curves for CoAs2O4 measured in 100 Oe in both alignment configurations (along crystal c-axis, and perpendicular to it). In the inset, it was shown a corresponding high-field measurement (10 kOe). | ||
4. Conclusions
CoAs2O4 and NiAs2O4 were synthesised using low-temperature hydrothermal method, which resulted in beautiful, pure and homogeneous single crystals. In this manner, among isotypic M2+X23+O4 compounds, the measuring of the magnetic properties using single-crystals was possible for the first time. Single-crystals also made us possible to extend our study of magnetic and vibrational properties to anisotropy measurements. MX2O4 compounds could be obtained by different synthesis routes (flux method, solid-state reaction, high-temperature hydrothermal method), but with the exception of CuAs2O4, low-temperature hydrothermal method was used for the crystallization of suitable M2+X23+O4 material for the first time.The structurally characterised single crystal of NiAs2O4, exhibit magnetic properties in accordance with the reported data. The magnetic susceptibility of NiAs2O4 at TN = 53 K in high magnetic field of 10 kOe shows behaviour consistent with a transition to the antiferromagnetic ground state. However, at 20 K another transition to the ferromagnetic state has been clearly observed, which might be attributed to the uncompensated surface spins due to Ni2+ magnetic moments that are not coordinated in the same way as expected for the title compound. The SQUID measurement of the single crystal of CoAs2O4 reveals some subtile interplay between AFM and FM interactions in the system as evidenced as FM-like transition at 105.5 K in small magnetic field and AFM-like transition in 10 kOe and above. The transition at 105.5 K is, according to our knowledge, the highest critical temperature among all similar structures.
Acknowledgements
The authors gratefully acknowledge financial support from the Austrian Science Foundation (FWF) (Grant no. V203-N19), Austrian Student Exchange Service (ÖAD) and Austrian Ministry of Science (BM.W_f) (Bilateral cooperation project with Croatia 2014–15, Grant no. HR05/2014). The authors are thankful to Andreas Artač, M.Sc and Prof. Dr Eugen Libowitzky for assisting during spectroscopic analysis.References
- S. Lösel and H. Hillebrecht, Z. Anorg. Allg. Chem., 2008, 634, 2299–2302 CrossRef.
- F. Pertlik, TMPM, Tschermaks Mineral. Petrogr. Mitt., 1975, 22, 211–217 CrossRef CAS.
- S. Ghose, P. K. S. Gupta and E. O. Schlemper, Am. Mineral., 1987, 72, 629–632 CAS.
- C. Giroux-Maraine and G. Perez, Rev. Chim. Miner., 1975, 12, 427–432 CAS.
- H. Fjellvåg and A. Kjekshus, Acta Chem. Scand., Ser. A, 1985, 39, 389–395 CrossRef PubMed.
- R. Fischer and F. Pertlik, TMPM, Tschermaks Mineral. Petrogr. Mitt., 1975, 22, 236–241 CrossRef CAS.
- J. R. Gavarri, R. Chater and J. Ziółkowski, J. Solid State Chem., 1988, 73, 305–316 CrossRef CAS.
- B. P. De Laune and C. Greaves, J. Solid State Chem., 2012, 187, 225–230 CrossRef CAS PubMed.
- E. G. Puebla, E. G. Ríos, A. Monge and I. Rasines, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 2020–2022 CrossRef.
- R. Chater, J. R. Gavarri and A. W. Hewat, J. Solid State Chem., 1985, 60, 78–86 CrossRef CAS.
- H. T. Witteveen, Solid State Commun., 1971, 9, 1313–1315 CrossRef CAS.
- F. Pertlik, Monatsh. Chem., 1988, 119, 4581 Search PubMed.
- SAINT, Bruker AXS Inc., 5465 East Cheryl Parkway, Madison, WI, USA, 53711–5373, 2000 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- K. Robinson, G. V. Gibbs and P. H. Ribbe, Science, 1971, 172, 567–570 CAS.
- M. E. Fleet, Mineral. Mag., 1976, 40, 531–533 Search PubMed.
- M. Wildner, Z. Kristallogr., 1992, 202, 51–70 CrossRef CAS PubMed.
- F. Emmerling and C. Röhr, Z. Naturforsch., B: J. Chem. Sci., 2003, 58, 620–626 CAS.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1988, 47, 192–197 CrossRef.
- S. Bahfenne, L. Rintoul and R. L. Frost, Am. Mineral., 2011, 96, 888–894 CrossRef CAS.
- A. G. Nord, P. Kierkegaard, T. Stefanidis and J. Baran, Chem. Commun., 1988, 1–40 Search PubMed.
- L. Bencivenni and K. A. Gingerich, J. Mol. Struct., 1983, 99, 23–29 CrossRef CAS.
- R. L. Frost and S. Baffenne, J. Raman Spectrosc., 2010, 41, 325–328 CAS.
- N. W. Ascroft and N. D. Mermin, Solid State Physics, Saunders College Publishing, USA, 1976, pp. 657–658 Search PubMed.
- L. J. De Jongh, Magnetic properties of layered transition metal compounds, Kluwer Academic Publishers, Netherland, 1990 Search PubMed.
- F. Bodker, M. F. Hansen, C. Bender Koch and S. J. Morup, J. Magn. Magn. Mater., 2000, 221, 32–36 CrossRef CAS.
- M. Jagodič, Z. Jagličić, A. Jelen, J.-B. Lee, Y.-M. Kim, H. J. Kim and J. Dolinšek, J. Phys.: Condens. Matter, 2009, 21, 215302 CrossRef PubMed.
- H. Shim, P. Dutta, M. S. Seehra and J. Bonevich, Solid State Commun., 2008, 145, 192–196 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CIF files of the structures. See DOI: 10.1039/c4ra16122j |
| This journal is © The Royal Society of Chemistry 2015 |