
Aryl-substituted symmetrical and unsymmetrical benzothiadiazoles†
Abstract
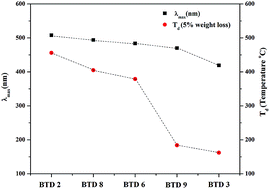
A set of benzothiadiazoles (BTD) of the type D–π–A–π–D and D1–π–A–π–D2 were designed and synthesized by the Pd-catalyzed Sonogashira cross-coupling reaction. Their photophysical and electrochemical properties were studied. The substitution of anthracene on BTD improves its thermal stability, and lowers the HOMO–LUMO gap, which results in a red shift of the electronic absorption. The experimental optical gap values show good agreement with the calculated HOMO–LUMO gap.
 Please wait while we load your content...
Please wait while we load your content...

