
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile ultrasonication approach for the efficient synthesis of ethylene glycol-based thermoresponsive nanogels
Catalina
Biglione†
a,
Ana
Sousa-Herves†
b,
Martina
Menger
b,
Stefanie
Wedepohl
b,
Marcelo
Calderón
*b and
Miriam C.
Strumia
*a
aLAMAP Laboratorio de Materiales Poliméricos, IMBIV-CONICET, Departamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Haya de la Torre y Medina Allende, X5000HUA Córdoba, Argentina. E-mail: mcs@fcq.unc.edu.ar
bInstitut für Chemie und Biochemie, Freie Universität Berlin, Takustrasse 3, 14195 Berlin, Germany. E-mail: marcelo.calderon@fu-berlin.de
First published on 26th January 2015
Abstract
The employment of nanogels (NGs) for drug delivery purposes has experienced a huge increase during the last decades. Among the different NGs, those displaying stimuli-responsive properties are of special interest. In particular, NGs that are able to swell or shrink by the action of temperature are very promising materials for applications in the biomedical and biological fields. In this work we present the preparation of thermoresponsive ethylene glycol-based NGs employing a simple and reliable ultrasonication approach. By this means, the reaction times could be shortened and the NGs formation could be performed even at room temperature and in the presence of oxygen. The NGs have been characterized by different techniques and their cloud point could be tuned by changing the molar ratio between the monomers. Finally, the NGs were labelled with Rhodamine B and their cellular uptake and cytotoxicity have been analysed.
Introduction
During the last decades, the employment of nanogels (NGs) as drug delivery systems has received increasing attention due to their unique chemical and physical versatility. NGs are a type of cross-linked, nanometric-sized hydrogel particles that offer several advantages over other polymer-based drug delivery systems.1–3 Thus, they can be easily synthesized with tunable sizes on the nanometer scale and present a higher stability upon dilution compared to other drug delivery systems such as classical polymeric micelles. NGs have a large surface area that allows further modification with biomolecules or targeting ligands and the interior network structure can be employed for the encapsulation and protection of drugs, proteins, or nucleic acids.1,4One of the most interesting features of NGs is their ability to undergo rapid conformational changes in response to an external stimulus such as pH, temperature, reductive environments, magnetic fields, or irradiation.3,5–7 In particular, NGs prepared from thermoresponsive polymers exhibiting a lower critical solution temperature (LCST) in aqueous medium are promising materials for bioapplications such as temperature-induced drug delivery or tissue engineering.5,8–12 Such polymers are soluble in water below the LCST but collapse at temperatures above it, prompting the expulsion of water and the consequent shrinking of the NG. Hence, temperature can be used as a simple external stimulus for controlling the structural shape of these NGs, trigger drug release, or to improve cellular targeting.13,14 Poly(N-isopropylacrylamide) (PNIPAM) is one of the most studied thermoresponsive polymers,6,8,15,16 with a LCST of 32 °C, but its application in the biomedical field has been hampered by its strong hysteresis of the thermal solubility transition,17,18 significant influence of end groups on the thermal behaviour,19 and poor biocompatibility profile.20
As a more biocompatible alternative, Lutz and coworkers have recently described the thermoresponsive behaviour of copolymers of 2-(2-methoxyethoxy)ethyl methacrylate (MEO2MA) and oligo(ethylene glycol) methacrylate (OEGMA).21–25 Such copolymers respond to temperature changes in an extent comparable, and in some cases superior, to that of PNIPAM. In addition, the phase transitions of (MEO2MA-co-OEGMA) copolymers were found to be completely reversible and relatively insensitive to important parameters such as copolymer concentration, ionic strength, or chain-length.22 Hu and coworkers have exploited the thermosensitive behaviour of (MEO2MA-co-OEGMA) copolymers for the preparation of temperature-responsive microgels.26,27 To that aim, a free radical polymerization and precipitation approach at high temperatures (70 °C) and relatively long reaction times (6 h) was employed. The monomers were initially dispersed in water and, once the copolymers were formed, they precipitated at a temperature above the LCST yielding to the formation of particles with micrometer sizes.26
Herein, we present a novel, reliable, easier, and faster methodology for the preparation of biocompatible thermoresponsive NGs with potential applications in the biomedical field. We have employed ethylene glycol-based MEO2MA, OEGMA, and 2-hydroxyethyl ether methacrylate (HEMA) as monomers and tetraethylene glycol dimethacrylate (TEGDMA) as the crosslinker (Fig. 1). Notably, HEMA has been incorporated into the NGs because its functionality allows the further covalent modification of the NGs with other relevant molecules, i.e., targeting ligands, dyes, or drugs.28 Our strategy is based on the use of ultrasonication to template NGs formation. This approach has allowed the decrease of reaction times in a high extent compared to typical methodologies for preparing NGs such as free radical dispersion/precipitation polymerization technique.29 We hypothesize that the shortened reaction times can be attributed to a higher monomers/crosslinker concentration in the nanodroplets formed during the ultrasonication process. In addition, NGs synthesis could be performed even at room temperature (rt). This is of special relevance for the encapsulation of sensitive biomolecules (such as proteins or nucleic acids) that may lose their activities at the high temperatures typically needed for the polymerization precipitation approach. Moreover, our strategy allows the formation of the NGs without the need of employing an inert atmosphere.26 The combination of those features, i.e., shorter reaction times, rt, and experimental simplicity, results in an attractive strategy for the formation of biocompatible, well-defined thermoresponsive NGs.
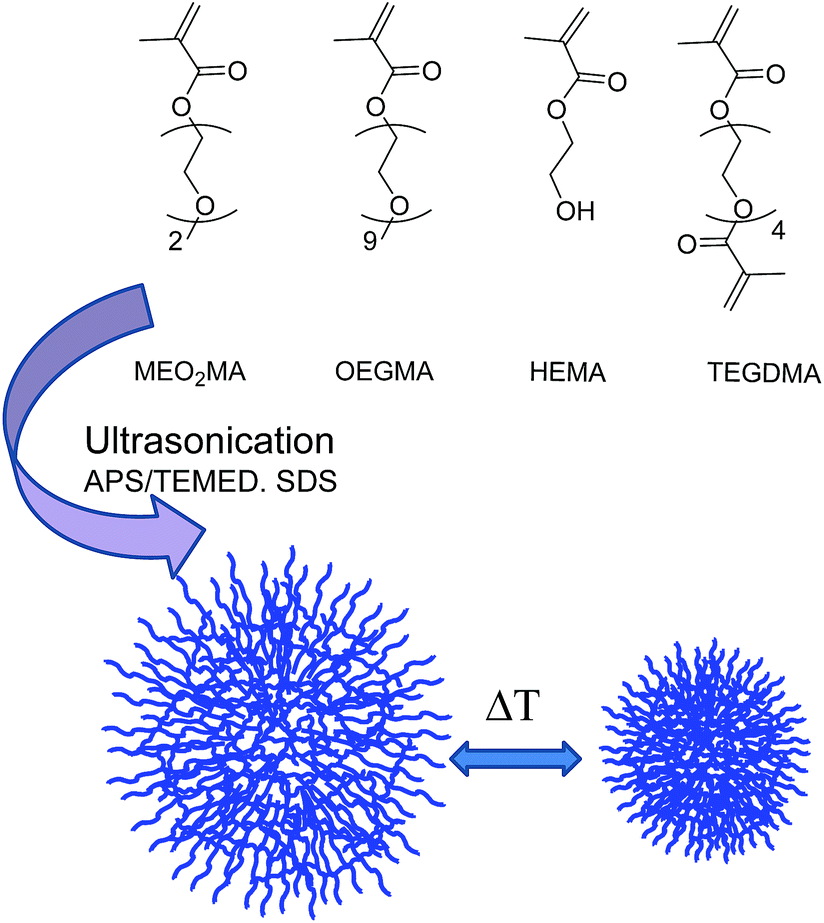 | ||
| Fig. 1 Schematic representation of thermoresponsive NGs. | ||
Experimental
Materials and methods
MEO2MA, OEGMA (Mn 475 g mol−1), HEMA, TEGDMA, tetramethylethylenediamine (TEMED), and Rhodamine B (RhB) were purchased from Sigma Aldrich. Sodium dodecyl sulfate (SDS) was obtained from Biopack. Ammonium persulfate (APS) was purchased from Anedra. Purification by dialysis was performed with membranes of cellulose ester (MWCO 50 KDa; SpectrumLabs). IR spectra were measured in a Nicolet 5SXC spectrometer. 1H NMR spectra were recorded on a Brüker Avance II (400 MHz). NMR chemical shifts are reported in ppm (δ units) downfield from the CDCl3 signal.General method for the synthesis of nanogels
NGs synthesis was performed with an ultrasonication device, Omnic Ruptor 4000. Several parameters, such as reaction times and the amount of crosslinker, surfactant, and initiator, were assessed in order to optimize the formation of the NGs. Once these conditions were experimentally determined, we prepared several NGs with different ratios between the monomers to fine tune their response towards temperature (Table 1). In a representative example for NGs synthesis (NG 10 in Table 1), the monomers MEO2MA (169 mg, 0.9 mmol), OEGMA (47.5 mg, 0.1 mmol), HEMA (4%), and the crosslinker TEGDMA (1.5%) were dissolved in distilled water. Then, the surfactant SDS (1.6%), the initiator, APS (1.6%), and TEMED (0.3 mL, 0.25 M) were added to the aqueous solution. All the percentages were calculated from the total moles of monomers. Subsequently, 10 mL of distilled water were added and the solution was heated to 60 °C. The reaction mixture then was ultrasonicated discontinuously (6 times) at 70% of power during the reaction. Since the amount of SDS was kept below its critical micelle concentration, it is expected to act as a surfactant stabilizing the nanodroplets formed in the ultrasonication process. Finally, the resulting NGs were extensively purified by dialysis (MWCO 50 kDa) in water (3 days, rt) and stored at 4 °C.| Nanogel | Molar ratio MeO2MA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :OEGMA :OEGMA |
HEMA | Temp. | Time (min) |
|---|---|---|---|---|
| NG 1 | 6:4 |
4% | 60 °C | 15 |
| NG 2 | 7.5:2.5 |
4% | 60 °C | 15 |
| NG 3 | 7:3 |
4% | 60 °C | 30 |
| NG 4 | 7:3 |
4% | 60 °C | 15 |
| NG 5 | 8:2 |
4% | 60 °C | 30 |
| NG 6 | 8:2 |
4% | 60 °C | 15 |
| NG 7 | 8:2 |
4% | 40 °C | 15 |
| NG 8 | 8:2 |
8% | 60 °C | 15 |
| NG 9 | 8:2 |
4% | rt | 15 |
| NG 10 | 9:1 |
4% | 60 °C | 15 |
Nanogels characterization
All NGs were characterized by IR and 1H NMR. The size of the NGs in aqueous solution was measured by dynamic light scattering (DLS). The measurements were carried out by a Malvern Zetasizer instrument with a He–Ne laser (λ = 633 nm) and scattering angle of 173°. Prior to the measurements, all samples were filtered through a 0.45 μm pore size filter. Atomic force microscopy (AFM) was employed to confirm the spherical shape of the NGs. The AFM tapping mode images were recorded in the air under ambient conditions, with a MultiMode 8 AFM equipped with a Nanoscope V controller from Veeco Instruments, Santa Barbara, California. 30 μL of a 0.1 mg mL−1 NG solution were deposited into cleaved mica and kept for about 3 min. After that time, the liquid was carefully removed with filter paper. Statistical analyses were performed with ImageJ software in a region of 5 μm × 5 μm.Analysis of the thermoresponsive behaviour of the NGs
The responsive behaviour of the NGs towards temperature was analysed by UV spectroscopy, DLS, and Nanoparticle Tracking Analysis (NTA). Cloud points (Cp) were measured on a Cary 100 Bio UV-Vis spectrophotometer equipped with a temperature-controlled, 6 position sample chamber. NGs water solutions were heated at 0.2 °C min−1 while monitoring both the transmittance (%T) at 500 nm (1 cm path length) and the solution temperature (from 23 to 70 °C), as determined by the internal temperature probe. Cp of each NG was determined using the minimum of the first derivate of graph %T vs. temperature. 3 cooling–heating cycles were performed for each measurement. DLS measurements were performed at 25 °C and 60 °C to determine the size of the NGs below and above their Cp. In addition, for one of the NGs, the correlation between the nanogel size in solution and the temperature (ramp from 25 to 65 °C, 2 measurements every 0.5 °C) was studied with DLS. NTA measurements were performed in a Nanosight NS500 (Malvern) and employed to determine the size and concentration of the NGs. Samples were prepared by diluting 100 times with water the solutions prepared for DLS measurements and immediately measured at 25 and 55 °C (55 °C is the maximum operating temperature). Particle sizes and concentration are given as the average of 3 measurements.In vitro cell viability studies
A549 lung cancer cells (#ACC-107) were obtained from DSMZ (Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures). Cells were routinely maintained in DMEM with 10% FCS (Biochrom AG) and 1% penicillin/streptomycin (Life Technologies) at 37 °C and 5% CO2.To assess cell viability using the MTT test, 10000 cells per well were seeded in 96-well plates in 100 μL of medium. After attaching overnight, medium was removed and replaced with 50 μL fresh medium and 50 μL of medium containing different concentrations of NGs. After 48 h incubation at 37 °C and 5% CO2, the cell culture supernatant was discarded and the cells were washed twice with 200 μL PBS per well. 100 μL per well fresh medium and 10 μL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT, 5 mg mL−1 stock solution in PBS) were added and incubated for another 4 h at 37 °C. The supernatant was then discarded and replaced with 100 μL per well 0.04 N HCl/2-propanol solution to dissolve the formazan crystals. Absorbance was read at 570 nm in a Tecan Infinite M200 Pro microplate reader. Each sample was measured in triplicate and 3 independent replications were performed. Calculation of half maximal inhibitory concentrations (IC50) values was done with GraphPad Prism.
NGs modification with RhB
NGs were covalently labelled with the dye RhB via carbodiimide-mediated ester formation between NG's OH groups and the carboxylic acid group of RhB. NG 8 was chosen due to the higher amount of HEMA on its composition. NG 8 (7.5 mg) was dissolved in dry DMF (200 μL). Separately, RhB (2.01 mg, 4.20 μmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 1 mg, 6.44 μmol), and 4-dimethylaminopyridine (DMAP, 0.642 mg, 5.25 μmol) were dissolved in 300 μL of DMF and stirred for 10 min. Afterwards, both solutions were mixed and allowed to stir overnight. Then, the labelled-NGs were purified by dialysis (MWCO 50 kDa) in MeOH/H2O (1:1, 3× solvent exchange) and in H2O (5 days) until no more free dye could be observed. NGs were freeze-dried (7.5 mg) and the amount of dye (0.46 μg per mg of NG) was determined by UV-Vis spectroscopy (MeOH, 1 mg mL−1, λmax = 561 nm, ε = 1.15 × 105 M−1 cm−1).
Cellular internalization studies
5000 cells per well were seeded onto cover slips in 24-well plates and grown overnight. The next day, the medium was discarded, washed twice with PBS, and replaced with 1 mL fresh medium (without phenol red). 10 μL of the RhB-labelled NG 8 (10 mg mL−1 in H2O) was added to the wells and incubated for 4 h at 37 °C and 5% CO2. The cells were then washed twice with PBS and fixed in 10% neutral buffered formalin for 20 min. After washing twice with PBS, the fixed cells were permeabilized with 1% Triton X-100 in PBS for 5 min and washed again 3 times. The samples stained for early endosomes were incubated with FITC conjugated anti-EEA1 antibody (BD Biosciences) 1:50 in PBS for 1 h. After 3 times washing with PBS, the samples were stained with DAPI (Carl Roth, 2.5 μg mL−1 in PBS) for 30 min. After 3 times washing with PBS, the cover slips were briefly dipped in H2O and mounted on microscope slides with ProTaqs® MountFluor mounting medium (Quartett GmbH). Images were recorded on a Leica SP8 confocal laser scanning microscope with LASAF Software (Leica).
Results and discussion
Synthesis and characterization of the NGs
Several parameters, such as reaction times, temperature, MeO2MA:OEGMA ratio, and HEMA concentration, have been tested in order to obtain the desired thermoresponsive NGs (Table 1). Firstly, different reaction times were screened (15 min, 30 min, 1 h, 1.5 h, 2 h, and 3 h) while the amount of monomers and crosslinker was kept constant (data not shown). The progress of NG formation with the desired nanometric sizes was determined by DLS, showing the presence of NGs with narrow size distributions even at short reaction times (15 and 30 min). After NGs purification, IR and 1H NMR spectroscopies confirmed the disappearance of the acrylate groups proving the formation of the polymeric network even for the shortest time tested (15 min). Therefore, 15 and 30 min were considered to be optimal reaction times and were employed in further studies. It should be noted that the reaction time could be decreased from 6 h, as previously reported in the literature,26 to only 15 min. The effect of the temperature during the NGs' formation was also explored (60 °C, 40 °C, and rt). Interestingly, we observed that the polymerization was complete, even when the NG synthesis was performed at rt (NG 9, Table 1). We hypothesize that the concentration of monomers and crosslinker in the nano-compartments formed during the ultrasonication process led to a faster polymerization, even with the presence of oxygen and without the need of heat.
Synthesized NGs were characterized by NMR, IR, and DLS. 1H NMR spectrum proved the purity of the NGs showing the complete disappearance of the vinyl protons (5.0–6.5 ppm). The IR spectrum confirmed the lack of the band corresponding to the double bond conjugated with carbonyl group between 1685–1665 cm−1. In addition, the presence of the characteristic bands at 2925 cm−1 and 2869 cm−1 (C–H aliphatic stretching) and at 1729 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, esters) verified the chemical structure of the NGs (Fig. 2).
O stretching, esters) verified the chemical structure of the NGs (Fig. 2).
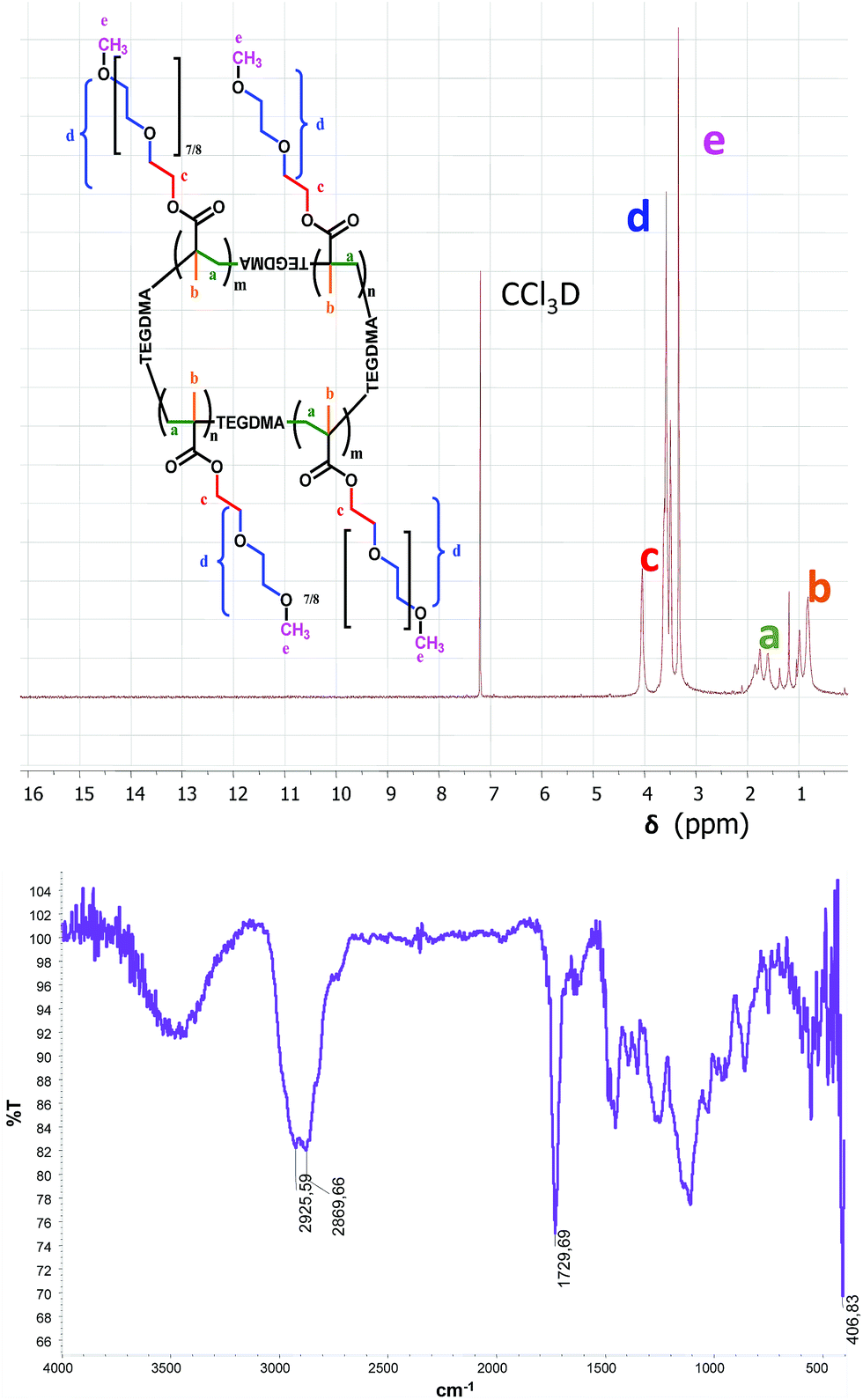 | ||
| Fig. 2 1H NMR (above) and FT-IR (below) of NG 8. | ||
DLS was employed to determine the size of the NGs in aqueous solution (Fig. 3a). All NGs sizes were in the range of 71–180 nm (at 25 °C), with PDI values around 0.1–0.2 in most of the cases (Table 2). The spherical shape of the NGs could be visualized by AFM (Fig. 3b, NG 3). As previously observed for other hydrophilic NGs,7,30 the size determined by AFM in dry state was slightly smaller (132 ± 20 nm) than the one measured in solution by DLS (143 nm).
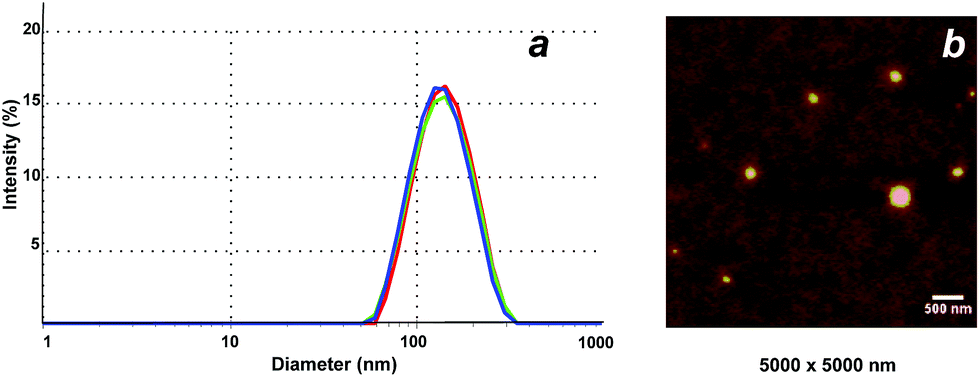 | ||
| Fig. 3 (a) DLS intensity size distribution (3 measurements) and (b) AFM image of NG 3. | ||
| Sample | Molar ratio MeO2MA:OEGMA |
HEMA | Cp | Size (nm) 25 °C below Cp | PDI 25 °C | Size (nm) 60 °C above Cp | PDI 60 °C |
|---|---|---|---|---|---|---|---|
| NG 1 | 6:4 |
4% | 62.0 | 71 | 0.358 | 314 | 0.564 |
| NG 2 | 7.5:2.5 |
4% | 50.3 | — | 1 | 528 | 0.07 |
| NG 3 | 7:3 |
4% | 57.3 | 143 | 0.147 | 353 | 0.173 |
| NG 4 | 7:3 |
4% | 57.4 | 128 | 0.224 | 250 | 0.29 |
| NG 5 | 8:2 |
4% | 49.2 | 145 | 0.159 | 229 | 0.02 |
| NG 6 | 8:2 |
4% | 47.9 | 114 | 0.141 | 173 | 0.22 |
| NG 7 | 8:2 |
4% | 44.0 | 156 | 0.23 | 297 | 0.14 |
| NG 8 | 8:2 |
8% | 52.5 | 149 | 0.136 | 246 | 0.104 |
| NG 9 | 8:2 |
4% | 46.7 | 174 | 0.18 | 643 | 0.737 |
| NG 10 | 9:1 |
4% | 37.7 | 140 | 0.194 | 519 | 0.08 |
It could be observed that the size of the NGs increased with the amount of the co-monomer HEMA. Thus, comparing NGs with the same MEO2MA:OEGMA molar ratio, it was shown that NG 6 (4% HEMA) was 114 nm in size, while NG 8 (8% HEMA) had a size of 181 nm. Furthermore, in general terms, an increased concentration of the hydrophilic monomer resulted in a larger size of the NGs. This tendency is not observed for NG 10, presumably due to a less homogeneous NG's structure. The same trend was observed for NGs with an increased reaction time. As an example, NG 5 (30 min reaction time) was approx. 30 nm larger than NG 6 (same composition, 15 min reaction time). Finally, the reaction temperature appears to have an influence on the size of the nanogels: the higher the temperature, the smaller the NGs (NGs 6, 7, and 9, Table 2).
Thermoresponsive properties of the NGs
Measurements of transmittance (%T) of UV-Vis light (λ = 500 nm) from aqueous solutions of NGs were performed to determine the Cp as an indication of their thermoresponsive behavior. It has been reported that LCST value of the co-polymers prepared from MEO2MA and OEGMA can be adjusted by varying the copolymer composition.25 The LCST of pure MEO2MA polymer is 26 °C, whereas the LCST of pure OEGMA polymer is 93 °C.25 In order to fine tune the Cp of the NGs, we decided to explore different molar ratios between the monomers OEGA and MEO2MA (from 9:1 to 6:4, Table 1). By this means, the Cp could be varied from 62.0 °C to 37.7 °C. As expected, the Cp increased when the amount of Me2OMA was decreased. Thus, NG 1, prepared from a 6:4 MeO2MA:OEGMA molar ratio, presented the highest Cp value (62 °C) while NG 10, synthesized from a 9:1 MeO2MA:OEGMA molar ratio showed the lowest Cp value (37.7 °C). Such Cp values are of particular interest for some applications in cancer therapy such as tumor ablation, in which temperatures above 45 °C are employed.31 Nevertheless, the NGs prepared with the highest or lowest MeO2MA:OEGMA molar ratios seemed to be the less homogeneous formulations. The effect of HEMA concentration was also explored. The NG with a higher amount of HEMA (8%, NG 8) showed a higher Cp than the corresponding counterpart prepared with 4% of co-monomer (NG 6), presumably due to the hydrophilic nature of HEMA. The normalized UV curves for selected NGs (NG 3 and 8) obtained measuring the %T vs. temperature for representative NGs are depicted in Fig. 4. Notably, as shown in Fig. 4, the thermoresponsivity of the NGs was found to be reversible at least for 3 temperature cycles with almost no sign of hysteresis.
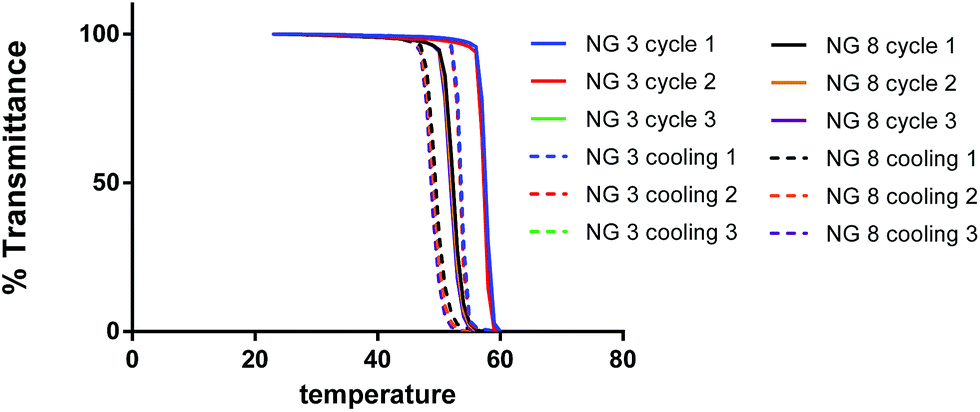 | ||
| Fig. 4 Normalized curves of %T vs. temperature of 3 heating–cooling cycles for NGs 3 and 8. | ||
DLS could also be employed for proving the swelling–shrinking behaviour of the NGs in response to an increase in the temperature. Fig. 5a and Table 2 show the difference in the hydrodynamic diameter below and above the Cp (i.e. 25 °C and 60 °C). Above the Cp, the NGs turned turbid (Fig. 5b) and showed a marked increase in their hydrodynamic sizes. This observation should be attributed to the shrinking and aggregation of the smaller and more hydrophobic NGs above their Cp. To further prove this hypothesis, we performed a DLS measurement for NG 8 using a temperature ramp from 25 to 65 °C. The results showed a strong aggregation for the NGs at temperatures above their Cp. In addition, in order to confirm this premise, NTA measurements at 25 and 55 °C were performed for NG 8. NTA showed that the number of particles decreased from 9.1 × 107 particles per mL to 1.6 × 107 particles per mL when temperature was increased from 25 to 55 °C. The size obtained by NTA increased from 227 ± 63 nm at 25 °C to 297 ± 113 nm at 55 °C.
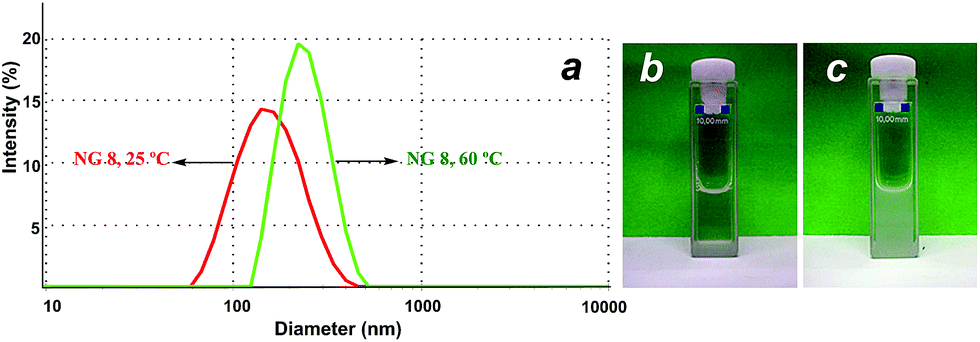 | ||
| Fig. 5 (a) DLS size histogram of NG 8 at 25 °C (left curve) and 60 °C (right curve). (b) Image of NG 8 at rt and (c) at 60 °C. | ||
Cytotoxicity of the NGs
In order to study if the NGs had any toxic effect on cells in vitro, A549 lung cancer cells were incubated with NG 1, NG 5, NG 8, and NG 10 for 48 h. Fig. 6 shows the relative cell viabilities of A549 cells at different NG concentrations as determined by MTT test. IC50 values as calculated by nonlinear dose–response curve fit (not shown) were as high as about 0.3 mg mL−1 for NGs 5 and 10, and 1.2 mg mL−1 for NGs 1 and 8. As can be observed, the most hydrophilic NGs, i.e., those having the least amount of MeO2MA (NG 1) or the highest amount of HEMA (NG 8), had less effect on cell viability than the more hydrophobic ones. Overall, the NGs caused no severe reduction in cell viability below 50% up to a concentration of 0.1 or 1 mg mL−1, respectively, showing the low toxicity of the ethylene glycol-based NGs. | ||
| Fig. 6 Cell viability as determined by MTT test on A549 cells after exposure of different concentration of NGs. | ||
Cellular internalization studies
The internalization of RhB-labelled NG 8 by A549 cells was investigated. In Fig. 7, a confocal laser scanning microscopy image of A549 cells incubated with RhB labelled NG 8 is shown.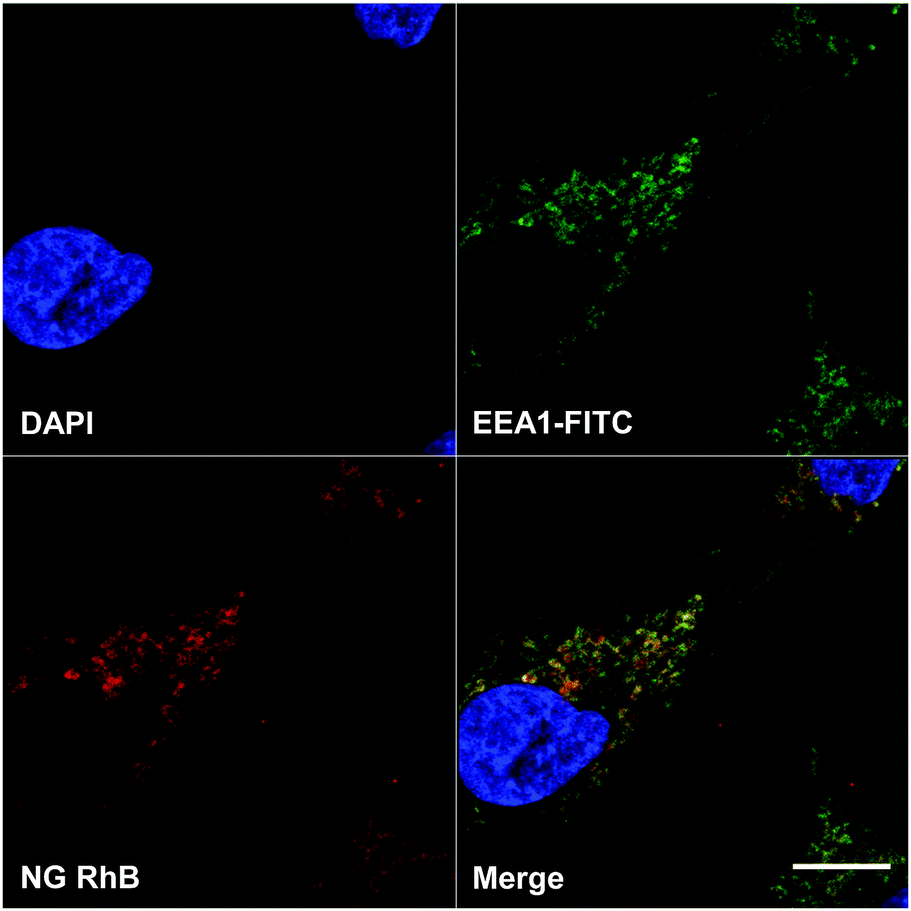 | ||
| Fig. 7 Confocal laser scanning microscopy image of A549 cells incubated with RhB labelled NG 8. Blue: nuclei, green: early endosomes marker EEA1, red: RhB. Scale bar: 10 microns. | ||
In the single channel images, the blue colour represents the cell nuclei and the early endosome marker EEA1 is shown in green. In red, RhB label of the NGs can be clearly seen in compartments and structures inside the cells, partly colocalizing with the endosome marker, which resulted in the yellow colour of the merged channels image.
With this feature, the ability to enter into cancer cells, the prepared NGs demonstrate their potential to be applied as drug delivery systems.
Conclusions
In conclusion, we have described a novel, simple, and fast method based on ultrasonication for the preparation of ethylene glycol-containing thermoresponsive NGs. This methodology has allowed the polymerization reaction to perform at very short times, at rt, and in the presence of oxygen. MEO2MA, OEGMA, and HEMA have been employed as monomers and TEGDMA as the crosslinker. Several parameters have been considered in order to optimize the NG synthesis, and reactions have been monitored by 1H NMR and IR spectroscopies. NGs have been characterized by DLS and AFM, showing the presence of spherical particles with sizes between 71–180 nm. The thermoresponsive behaviour of the NGs has been analysed by UV measurements and DLS. The Cp of the NGs could be fine-tuned by changing the ratio between monomers. Finally, the NGs were labelled with a dye and their cytotoxicity and cellular uptake has been analysed.The NGs described in this work presented ideal sizes and polydispersities, as well as good cytotoxicity and cell permeation profiles. They are therefore envisioned as attractive delivery systems for the controlled release of biomacromolecules, imaging agents, or low molecular weight anticancer drugs.
Acknowledgements
We would like to thank the financial support of CONICET, SECyT-UNC, and FONCYT. M. C. thanks the collaborative research center SFB1112 (Project 04), the Helmholtz Virtual Institute “Multifunctional Biomaterials for Medicine”, and the NanoMatFutur award (13N12561) for financial support. M. C. and M. S. thank the cooperative research project sponsored by BMBF-Mincyt (2013) and Conicet-DFG (2014). We acknowledge the focus area Nanoscale of the FU Berlin (http://www.nanoscale.fu-berlin.de). C. B. thanks CONICET for the fellowship granted for her PhD studies. A. S.-H. is grateful for a Marie Curie Intra-European Fellowship (Project 302717). M. M. thanks the PROMOS Program of the FU Berlin. Furthermore, we are grateful to Dr P. Winchester for proofreading the manuscript.Notes and references
- A. V. Kabanov and S. V. Vinogradov, Angew. Chem., Int. Ed., 2009, 48, 5418–5429 CrossRef CAS PubMed.
- R. T. Chacko, J. Ventura, J. Zhuang and S. Thayumanavan, Adv. Drug Delivery Rev., 2012, 64, 836–851 CrossRef CAS PubMed.
- M. Asadian-Birjand, A. Sousa-Herves, D. Steinhilber, J. C. Cuggino and M. Calderon, Curr. Med. Chem., 2012, 19, 5029–5043 CrossRef CAS PubMed.
- M. H. Smith and L. A. Lyon, Acc. Chem. Res., 2011, 985–993 Search PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- M. Molina, M. Giulbudagian and M. Calderón, Macromol. Chem. Phys., 2014, 215, 2414–2419 CrossRef CAS.
- M. Giulbudagian, M. Asadian-Birjand, D. Steinhilber, K. Achazi, M. Molina and M. Calderon, Polym. Chem., 2014, 5, 6909–6913 RSC.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2012, 42, 7468–7483 RSC.
- J. Ramos, A. Imaz, J. Callejas-Fernandez, L. Barbosa-Barros, J. Estelrich, M. Quesada-Perez and J. Forcada, Soft Matter, 2011, 7, 5067–5082 RSC.
- J. Bergueiro and M. Calderón, Macromol. Biosci., 2014 DOI:10.1002/mabi.201400362.
- T. Hoare, B. P. Timko, J. Santamaria, G. F. Goya, S. Irusta, S. Lau, C. F. Stefanescu, D. Lin, R. Langer and D. S. Kohane, Nano Lett., 2011, 11, 1395–1400 CrossRef CAS PubMed.
- Z. Shen, A. Mellati, J. Bi, H. Zhang and S. Dai, RSC Adv., 2014, 4, 29146–29156 RSC.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- Y. Hiruta, M. Shimamura, M. Matsuura, Y. Maekawa, T. Funatsu, Y. Suzuki, E. Ayano, T. Okano and H. Kanazawa, ACS Macro Lett., 2014, 3, 281–285 CrossRef CAS.
- J. C. Cuggino, C. I. Alvarez I, M. C. Strumia, P. Welker, K. Licha, D. Steinhilber, R.-C. Mutihac and M. Calderon, Soft Matter, 2011, 7, 11259–11266 RSC.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- X. Wang, X. Qiu and C. Wu, Macromolecules, 1998, 31, 2972–2976 CrossRef CAS.
- X. Wang and C. Wu, Macromolecules, 1999, 32, 4299–4301 CrossRef CAS.
- Y. Xia, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2006, 39, 2275–2283 CrossRef CAS.
- H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu and J. Hirvonen, Biomaterials, 2005, 26, 3055–3064 CrossRef CAS PubMed.
- N. Badi and J.-F. Lutz, J. Controlled Release, 2009, 140, 224–229 CrossRef CAS PubMed.
- J.-F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- J. F. Lutz, K. Weichenhan, Ã. Akdemir and A. Hoth, Macromolecules, 2007, 40, 2503–2508 CrossRef CAS.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- J.-F. Lutz and A. Hoth, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- T. Cai, M. Marquez and Z. Hu, Langmuir, 2007, 23, 8663–8666 CrossRef CAS PubMed.
- Z. Hu, T. Cai and C. Chi, Soft Matter, 2010, 6, 2115–2123 RSC.
- D. J. Siegwart, A. Srinivasan, S. A. Bencherif, A. Karunanidhi, J. K. Oh, S. Vaidya, R. Jin, J. O. Hollinger and K. Matyjaszewski, Biomacromolecules, 2009, 10, 2300–2309 CrossRef CAS PubMed.
- S. Kawaguchi and K. Ito, in Polymer Particles, ed. M. Okubo, Springer, Berlin, Heidelberg, 2005, vol. 175, pp. 299–328 Search PubMed.
- S. Höfl, L. Zitzler, T. Hellweg, S. Herminghaus and F. Mugele, Polymer, 2007, 48, 245–254 CrossRef.
- Y. Zhang, W. Zhang, C. Geng, T. Lin, X. Wang, L. Zhao and J. Tang, Prog. Nat. Sci., 2009, 19, 1699–1704 CrossRef.
Footnote |
| † Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |