
A DFT-D study of hydrogen adsorption on functionalized graphene
Abstract
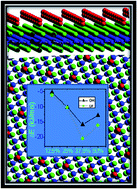
In this paper, we use density functional theory with dispersion correction functional (DFT-D) as implemented in the Vienna ab initio simulation package in order to investigate hydrogen adsorption on graphene (GH) and fluorographene (GF). The adsorption sites at different surface coverage rates were studied to determine the most stable configurations. The comparison between the results obtained using standard pure DFT functionals and dispersion corrected ones highlight the role of the dispersion effect in the adsorption energies and the orientation of the molecules relative to the surface. The coverage rate is found to increase up to 75% on the two sides, making these nanoporous materials promising candidates for hydrogen storage. Electronic properties such as density of states and band structures were calculated on both GH and GF systems. It is observed that after H2 adsorption the band gap of GH is only slightly modified, whereas the opposite trend is observed on GF.
 Please wait while we load your content...
Please wait while we load your content...

