
Cobalt supported on metal-doped ceria catalysts (M = Zr, Sn and Ti) for NO oxidation†
Yang Yuab,
Lei Zhongab,
Jie Dingab,
Wei Caiab and
Qin Zhong*ab
aSchool of Chemicsal Engineering, Nanjing University of Science and Technology, Nanjing 210094, P. R. China. E-mail: zq304@mail.njust.edu.cn; Fax: +86 25 84315517; Tel: +86 25 84315517
bNanjing AIREP Environmental Protection Technology Co., Ltd, Nanjing, Jiangsu 210091, P. R. China
First published on 25th February 2015
Abstract
A series of ceria oxides doped with 20 mol% of Zr, Sn and Ti were prepared by a citric sol–gel method throughout the thermal decomposition of the corresponding metallic propionates. Furthermore, cobalt oxides were loaded on the supports by a one-step sol–gel method. The results showed a solid solution when doping with Zr and Ti and SnO2 surface segregation in the case of Sn. We demonstrated that cobalt dispersion states were influenced by the doping metals. The Ce–Co metal-oxide catalyst possessed a higher amount of finely dispersed cobalt species, more oxygen vacancies and excellent redox ability. These features may be responsible for improving the catalytic activity of Ce–Co metal-oxides for NO oxidation.
1. Introduction
Development of methods to control NOx emission has been an active research area1 because NOx are associated with acid deposition, photochemical smog, and ozone depletion. The catalytic oxidation of NO to NO2 is a key step for many NOx removal techniques, such as fast selective catalytic reduction (fast SCR),2 NOx storage-reduction (NSR),3 continuously regenerating trap (CRT),4 and the wet scrubbing method.5 Therefore, many efforts have been focused on developing eligible catalysts for NO oxidation.6–10Ceria (CeO2) is one of the most reactive rare-earth oxides and attracts much attention due to its unique application in catalysts.6,8,9 The catalytic properties of ceria are considered to originate from the Ce4+/Ce3+ redox cycle. It is affected by various structural factors, such as composition modification, large surface area, preferential exposure of reactive facets and the oxygen vacancies concentration on its surface.11–14 However, it is well known that sintering effect often occurs when pure CeO2 is used at high temperature, resulting in the decrease of specific surface area and oxygen storage capacity (OSC).15 Many researches have been performed by incorporating foreign metal cations into the lattice of CeO2 to restrain this effect. These metal cations mainly contain three categories: (i) M2+ (Ca2+,16 Mg2+ (ref. 17)), (ii) M3+ (Al3+, Ga3+, In3+ (ref. 18 and 19)) and (iii) M4+ (Zr4+,6 Sn4+,20 Ti4+,21 Mn4+,22 Hf4+ (ref. 23)). Recently, our lab24 prepared a series of Cr/CexZr1−xO2 catalysts used for NO oxidation and found that the Zr-rich samples showed higher activity than Ce-rich sample; Dobrosz-Gómez et al.25 prepared a series of Au/CexZr1−xO2 used them in CO oxidation and the Au/Ce0.75Zr0.25O2 exhibited the best catalytic performance; Ayastuy et al.26 reported that Ce0.95Sn0.05O2 had the highest activity in CO oxidation reaction among the CexSn1−xO2 catalysts. According to the above-mentioned literatures, we could find that the previous works were mainly focused on investigating the catalytic property of the catalyst by modulating the ratio of Ce–M. However, there were almost few reports which focus on changing the doped metal cation of Co3O4/CexM1−xO2 for a comparative study.
It was widely reported that cobalt oxide based catalysts were potential candidate for NO oxidation. Wen et al. studied series of La1−xCexCoO3 perovskite oxide catalysts for nitrogen monoxide oxidation, and reported approximately 80% conversion on La0.8Ce0.2CoO3 at 300 °C.27 TiO2 and SiO2 supported Co3O4 catalysts were studied for NO oxidation by Irfan et al.28 They reported maximum of 69% conversion on Co3O4/SiO2 at 300 °C with high space velocity condition. Recently, Kim et al. studied NO oxidation on supported cobalt oxide synthesized by conventional wet impregnation method using various supports including CeO2, SiO2, ZrO2, TiO2 and SiO2, and reported that CeO2 supported catalyst exhibits better activity. They reported approximately 70% conversion at about 270 °C on Co-ceria and found that surface area of CeO2 played an important role on catalytic activity.29 As it was reported in the previous literatures, the support was crucial for the dispersion of active species, however, few reports discussed the different role of cobalt species in the process of NO oxidation.
In the present work, ceria doped with Zr4+, Sn4+, Ti4+ were used as supports to prepare cobalt-based catalysts and the obtained samples have been studied systematically by means of XRD, LRS, UV-vis DRS, TEM, N2-physisorption, H2-TPR, XRF, XPS and NO + O2 model reaction. The study was mainly focused on understanding the influence of dopant on the structure, texture, reduction, dispersion of cobalt species, oxygen vacancies, adsorption properties and catalytic performance of the obtained samples.
2. Experimental section
2.1. Preparation of the catalysts
The Ce0.8M0.2O2 (M = Zr4+, Sn4+ and Ti4+) solid solutions (Ce–M = 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) were prepared by sol–gel method. The Ce, Zr, Sn and Ti sources were Ce(NO3)3·6H2O, ZrOCl2, SnCl4 and TiCl4. The requisite quantity of Ce and M sources with citric acid (citric acid/(Ce + M) = 2) were added into distilled water to form solution. The above two solutions (Ce and M) together with citric acid were mixed together and stirred for 2 h. After that the mixture was heated at 90 °C under stirring until it became a viscous gel and dried at 120 °C for 12 h then the obtained solid was calcined in air at 500 °C for 4 h. In addition, pure CeO2 was prepared via the same procedure for comparison. These synthesized samples were CeO2, Ce0.8Zr0.2O2 (hereafter denoted as Ce–Zr), Ce0.8Sn0.2O2 (hereafter denoted as Ce–Sn), Ce0.8Ti0.2O2 (hereafter denoted as Ce–Ti) supports.
:1 molar ratio) were prepared by sol–gel method. The Ce, Zr, Sn and Ti sources were Ce(NO3)3·6H2O, ZrOCl2, SnCl4 and TiCl4. The requisite quantity of Ce and M sources with citric acid (citric acid/(Ce + M) = 2) were added into distilled water to form solution. The above two solutions (Ce and M) together with citric acid were mixed together and stirred for 2 h. After that the mixture was heated at 90 °C under stirring until it became a viscous gel and dried at 120 °C for 12 h then the obtained solid was calcined in air at 500 °C for 4 h. In addition, pure CeO2 was prepared via the same procedure for comparison. These synthesized samples were CeO2, Ce0.8Zr0.2O2 (hereafter denoted as Ce–Zr), Ce0.8Sn0.2O2 (hereafter denoted as Ce–Sn), Ce0.8Ti0.2O2 (hereafter denoted as Ce–Ti) supports.
The CoOx/S (S = CeO2, Ce–Zr, Ce–Sn, Ce–Ti) catalysts were prepared by one-step sol–gel method. The Co source was Co(NO3)2·6H2O. The Ce, M (M = Zr, Sn and Ti) and Co solution with citric acid (citric acid/(Ce + M + Co) = 2) were mixed together and stirred for 2 h. After that the mixture was heated at 90 °C under stirring until it became a viscous gel and dried at 120 °C for 12 h then the obtained solid was calcined in air at 500 °C for 4 h. These catalysts were denoted as Ce–Co, Ce–Zr–Co, Ce–Sn–Co and Ce–Ti–Co, respectively.
2.2. Sample characterization
The powder XRD patterns were recorded on a Beijing Purkinjie general instrument XD-3 X-ray diffraction using Cu-Kα radiation at 36 kV and 20 mA (2θ from 5° to 80°). The scanning speed is 8° min−1 and the step value is 0.04°. The average grain sizes of the catalysts were calculated using the Scherer's formula, eqn (1):
 | (1) |
 | (2) |
Laser Raman spectra (LRS) were recorded on a Renishaw Invia Raman Microscope with Ar+ radiation (514 nm). The laser light was focused onto the samples by using a microscope equipped with a 6100 objective lens.
Diffuse reflectance spectroscopy (DRS) was carried out on a Shimadzu UV-2550 UV-vis spectrophotometer. BaSO4 was the reference sample and the spectra were recorded in the range of 200–800 nm.
The micromorphology of the catalysts was examined on a JEOL JEM-2100 transmission electron microscope (TEM), and the sample was deposited on a copper mesh by means of dipcoating. The acceleration voltage is 200 kV.
Specific surface areas of the different catalysts were determined by N2 adsorption–desorption measurements at −196 °C by employing the Brunauer–Emmet–Teller (BET) method (Gold App V-sorb 2800 p), and the pore volume and pore size of the samples were calculated by Barrett–Joyner–Halenda (BJH) method. The systematic error calculated for BET surface area, pore volume and pore size was given with the accuracy at ±5 m2 g−1, ±5 mm3 g−1 and ±0.5 nm. Prior to N2 adsorption, the sample was outgassed at 200 °C for 12 h to desorb moisture adsorbed on the surface and inside the porous network.
Hydrogen temperature programmed reduction (H2-TPR) was performed in a quartz U-tube reactor on an automated chemisorption analyzer (Quantachrome Instruments) by the GC method. About 100 mg sample was pretreated in N2 stream at 600 °C for 0.5 h. As the sample was cooled downed to 50 °C, switched N2 to H2–N2 mixture gas (10% H2, v/v) at a flow rate of 70 mL min−1. H2-TPR was performed by heating the sample from 50 to 700 °C, at the same time, the consumption of H2 was detected by a thermal conductivity detector (TCD).
X-ray photoelectron spectra (XPS) was performed on a Thermo ESCALAB 250 (USA) apparatus with Al Kα X-rays (hv = 1486.6 eV) radiation operated at 150 W to investigate the surface atomic concentrations and the oxidation state distribution of the elements in the samples. The samples were compensate for charging with low-energy electron beam, and the peak of C 1s (binding energy = 284.4 eV) was used to correct for sample charging. This reference gave BE values with an accuracy ±0.1 eV. And the atomic surface ratios of the corresponding species were given with the accuracy at ±0.1%. The penetration depth of the XPS probe is 10 nm.
X-ray fluorescence spectra (XRF) were carried out on an FL3-TCSPC apparatus to determine the bulk composition of these catalysts. The X-ray tube was operated at 60 kV and 20 mA. The accurate measurement mode was adopted to determine the contents of Co, Ce, Zr, Sn and Ti elements of these catalysts.
2.3. Catalytic testing
The catalytic oxidation of NO was performed in a fixed-bed flow microreactor under atmospheric pressure. Typically, 300 mg sample (sieve fraction of 40–60 mesh) was placed in a quartz reactor (6.8 mm i.d.); the reactant gas mixture (390 ppm NO, 8% O2, N2 balance) was fed to the reactor with a total flow rate of 100 mL min−1, corresponding to a gas hour space velocity (GHSV) of 35400 h−1. The steady-state tests were conducted isothermally every 25 °C from 200 to 400 °C and the gas products (after 90 min reaction) were analyzed by a Ecom-JZKN flue gas analyzer (Germany). The NO conversion is defined as:| NO conversion = (NOin − NOout)/NOin × 100%. |
3. Result and discussion
3.1. Catalytic activity of ceria-based mixed oxides (NO oxidation)
The profiles for NO conversion as a function of reaction temperature over these catalysts were present in Fig. 1a and b. For the Co-containing samples (Fig. 1a), the sequence of catalytic activity expressed as NO conversion is Ce–Co > Ce–Zr–Co > Ce–Sn–Co > Ce–Ti–Co. In contrast, the pure CeO2 and CeO2–MOx (M = Zr4+, Sn4+ and Ti4+) samples were also prepared to evaluate the catalytic activity. Although the catalytic activity of all the supports is low, modified supports have relative higher activity than pure CeO2. In general, the Co-containing samples shows higher catalytic activity than the supports, indicating Co species play an important role in the NO oxidation process. | ||
| Fig. 1 Comparing NO conversion over the supports and catalysts. | ||
3.2. Structural characteristics (XRD, LRS, UV-vis DRS and TEM)
Fig. 2 shows the XRD patterns of these samples. It could be seen that all the diffraction peaks of these samples except Ce–Sn were characteristic of typical cubic fluorite phase of ceria (PDF-ICDD 34-0394). The characteristic lines attributed to ZrO2 and TiO2 were absent, the diffraction peaks of Ce–Zr and Ce–Ti shifted to high angle direction slightly and the peaks became wider compared to CeO2, which indicated that Zr4+ and Ti4+ were successfully doped into the lattice of CeO2 to form uniform solid solutions. As for Ce–Sn, the diffraction peak of SnO2 was observed, showing the absence of solid solution or little solid solution forming. In contrast, the Ce–Sn–Co sample showed no SnO2 peak and the (1 1 1) peak of CeO2 shifted to higher angle, this phenomenon indicated the adding Co could contribute to the forming of solid solution. The grain sizes of these samples are summarized in Table 1. The grain sizes of Ce–Zr, Ce–Sn and Ce–Ti were smaller than CeO2, suggesting that the introduction of Zr4+, Sn4+ and Ti4+ inhibited the crystal growth of the cubic phase.21 After introducing of cobalt, the grain sizes decreased slightly compared to their corresponding supports except for Ce–Ti–Co. This phenomenon was probably due to the incorporation of monolayer cobalt into the surface/subsurface layers of these supports.30 In addition, all the Co supported catalysts showed no peaks for crystalline cobalt oxides, indicating that cobalt oxide species were highly dispersed and/or present as small clusters and/or form solid solution which were difficult to be detected due to the limitation of XRD. | ||
| Fig. 2 XRD patterns of the synthesized samples. | ||
| Sample | Grain size (nm) | Position of Raman line (cm−1) | FWHM of Raman line (cm−1) | AII/AI |
|---|---|---|---|---|
| CeO2 | 16.4 | 464 | 9.6 | — |
| Ce–Zr | 7.7 | 470 | 52.3 | — |
| Ce–Sn | 15.4 | 464 | 15.5 | — |
| Ce–Ti | 5.5 | 432 | 69.9 | — |
| Ce–Co | 7.7 | 431 | 77.2 | 0.3113 |
| Ce–Zr–Co | 5.2 | 445 | 94.9 | 0.2098 |
| Ce–Sn–Co | 5.1 | 437 | 62.5 | 0.1327 |
| Ce–Ti–Co | 6.6 | 438 | 56.9 | 0.1024 |
Laser Raman spectroscopy is an intriguing tool to analyze the structural properties of oxide materials particularly nanostructured ceria-based solid solutions. The LRS of the supports are shown in Fig. 3a. The band at 464 cm−1 was the characteristic of the cubic fluorite structure of CeO2.31 The observed shift and broadening of the F2g peaks in the 464 cm−1 was attributed to the change in the M–O vibration which accounted for the difference in the ionic radius of the dopants.32 In our case, the observed decrease in the ceria particle size in the doped samples calculated by the Scherrer equation (Table 1) could agree with the shift and/or broadening of the F2g peak. The Raman lines assigned to MO2 (M = Zr4+, Sn4+ and Ti4+) were not observed. This results also confirmed that the doped cations entered the crystal lattice of CeO2 which was in line with XRD results.
 | ||
| Fig. 3 LRS of the supports (a) and the catalysts (b). | ||
For the catalysts, the Raman lines of cobalt species were absent in all of the samples. In addition, the main band of these catalysts were red or blue shift in contrast to their support. This phenomenon could be explained be two reasons. One is that the existence of an interaction between support and the active species.33 The other reason may be the incorporation of monolayer cobalt into the surface/subsurface layers of these supports, which was in consistent with XRD results.
Apart from the main peak of CeO2, a new band II appeared at ca. 570 to 630 cm−1, ascribing to the intrinsic oxygen vacancies.34 They were very essential for absorption/dissociation of oxygen molecule during the catalytic oxidation reactions, which in turn could affect the catalytic activity of the catalysts.35 Since the band II was related to oxygen vacancies, and the band I was the main band of these samples, the area ratio of AII/AI could reflect the concentration of oxygen vacancies. We could see from Table 1 that the Ce–Co had higher amount of oxygen vacancies.
UV-vis DRS technique is effectively used for the study of metal oxides to obtain information about the charge transfer transitions. Ceria exhibited strong absorption below 400 nm caused by charge transfer bands. DR spectra of these samples are presented in Fig. 4. Three characteristic bands of ceria corresponding to the O2− → Ce3+ and O2− → Ce4+ charge transfer bands and inter-band transitions in the wavelength range 250–340 nm were observed for all samples.36,37 The absorption band observed at ca. 470 nm can be attributed to the 1A1g → 1T1g forbidden d–d transitions of Co3+ in its octahedral sites.38 The peak observed at ca. 750 nm corresponded to Co2+ ions in its tetrahedral geometry.39 It was confirmed that the evidence of the Co3+ and Co2+ species from DRS study was in accordance with H2-TPR and XPS results.
 | ||
| Fig. 4 UV-vis spectra of these catalysts. | ||
TEM was used to investigate the morphology and microstructure of the sample. Fig. 5 shows the TEM and HR-TEM imagines of these catalysts. From TEM imagines, we could clearly see that the particles of all the catalysts were irregular. It could be seen from the HRTEM imagines that these irregular imagines were comprised of many small particles with a crystallite size of 3–5 nm, and there were clear voids with diameter 3–5 nm among the small particles, revealing the mesostructure of the CeO2 particles. Furthermore, the absence of Co-related particles' lattice in HRTEM images suggested a good dispersion of CoOx crystallites. From HRTEM, the interplanar distance of lattice fringes was 0.312 nm corresponding to (1 1 1) crystallographic plane of CeO2.40 In contrast, the fringes of Ce–M–Co (M = Zr4+, Sn4+ and Ti4+) were little lower than 0.312 nm, indicating the forming of Ce–M solid solution. The results were consistent with XRD and Raman results.
 | ||
| Fig. 5 TEM and HRTEM patterns of the samples. (A) Ce–Co; (B) Ce–Zr–Co; (C) Ce–Sn–Co; (D) Ce–Ti–Co. | ||
3.3. Textural characterization (N2-physisorption)
The N2 adsorption–desorption isothermal plots and the corresponding BJH pore size distribution curves of these samples were displayed in Fig. 6. It could be seen from Fig. 6a that all these samples were of classical type IV as defined by IUPAC and exhibited hysteresis loops of type H3, which indicated that these samples contained mesopores (2–50 nm) with narrow slit-like shapes.41 The pore size distribution curves suggested that all the samples had defined mesoporosities. The pore size at the maximum probability of the catalysts was 6.9, 11.3, 10.8 and 13.8 nm for Ce–Co, Ce–Zr–Co, Ce–Sn–Co and Ce–Ti–Co, respectively. The BJH pore size distribution plots of the various catalysts possessed nonuniform mesopore size distribution. | ||
| Fig. 6 N2 adsorption–desorption isotherms (a) and pore size distributions (b) of the samples. | ||
The BET surface area, pore volume and pore size of these catalysts are summarized in Table 2. Compared with Ce–Co, when the foreign metal cations were doped into the lattice of CeO2 (formed Ce–Zr, Ce–Sn, and Ce–Ti solid solutions), both the specific surface area and pore volume increased. These changes may be related to the grain size of the samples to some extent. In other words, the incorporation of Zr4+, Sn4+ and Ti4+ into the lattice of CeO2 could improve the texture property effectively. In order to explore the thermal stability, all of the four supports were calcined in air at 800 °C for 4 h in flowing air, and then, BET measurements were carried out, and the specific surface areas were summarized in Table S1.† Compared with pure CeO2, the degree of sintering (i.e. SBET(500) − SBET(800)/SBET(500)) was smaller when Zr4+, Sn4+ and Ti4+ incorporated into the lattice of CeO2. It indicated that the ability of anti-sintering of CeO2 was enhanced with the introduction of these foreign metal cations.
| Sample | Ce–Co | Ce–Zr–Co | Ce–Sn–Co | Ce–Ti–Co |
|---|---|---|---|---|
| BET (m2 g−1) | 21 | 46 | 65 | 39 |
| Pore size (nm) | 6.9 | 11.3 | 10.8 | 13.8 |
| Pore volume (mm3 g−1) | 66 | 192 | 188 | 257 |
3.4. Reduction properties of the catalysts (H2-TPR)
H2-TPR tests were performed to study the reducibility of these catalysts. Fig. 7 shows that the shape of these TPR profiles is very different, indicating the support has an important effect on the dispersion of Co3O4. With respect to the reduction steps of Co3O4, they were rather controversial in literature. Arnoldy and Moulijn42 observed a single step for the reduction of Co3O4, while many others43–45 reported that the reduction of Co3O4 was a two-step process, involving in the reduction to CoO. The results here clearly showed that the reduction behaviors of Co3O4 strongly depended on preparation, catalyst composition and dispersion behavior, consistent with the finding of Spadaro et al.46 Large particles of Co3O4 were often reduced directly to metallic Co in a single step, while fine particles of Co3O4 that interacted with ceria seemed to get reduced in two steps. The first step was generally promoted by component interaction between them, possibly ascribed to the lengthening in the Co–O bond, while the second step was perhaps delayed by the stabilization effect of ceria to cobalt ions, similar assignment has ever reported by Martínez-Arias et al. for CuO/CeO2 system.47 On the basis, the following assignments were made for the reduction of cobalt species in respect of the temperature ranges: | ||
| Fig. 7 H2-TPR profiles of these catalysts. | ||
Range I (below 320 °C): reduction of the surface adsorbed oxygen species (indicated as peak α for Ce–Co catalyst);
Range II (320–420 °C): reduction of Co3+ at the interface between Co3O4 and CeO2 to Co2+ (peak β);
Range III (420–500 °C): reduction of independent Co3O4 that weakly interacts with CeO2 directly to Co (peak γ);
Range IV (500–700 °C): reduction of Co2+ interacting with CeO2 to Co (peak θ).
Since the peak in Range II were related to the finely Co3O4 species and Range III were independent or bulk-like Co3O4 phase, we could infer the dispersion state of Co3O4 phase from the intensity of this peak. As shown the dotted box in Fig. 7, the peak area ratio of Aβ/Aγ was 1.8, 0.5, 0.4 and 0.3, referring to Ce–Co, Ce–Zr–Co, Ce–Sn–Co and Ce–Ti–Co, respectively. The reduction temperature of peak β and peak γ shifted to higher temperature in contrast with Ce–Co. Thus it could be seen that the support had significant effect on the reduction properties and component dispersion. Combined with the activities performance, we could infer that highly dispersed Co3O4 possessed better catalytic activity. Similar conclusions were reported by Luo et al.,13 they found that the finely dispersed CuO species had higher activity compared to bulk CuO species.
3.5. Bulk and surface analysis of the catalysts (XRF and XPS)
The bulk composition of these catalysts were characterized by XRF. As shown in Table 3, the chemical composition of the catalysts matched well the intended one, evidencing that the thermal decomposition of the propionates method allowed obtaining materials with a controlled stoichiometry. XPS was performed in order to illuminate the surface composition and the chemical state of the elements existing in the catalysts. The deconvoluted photoelectron spectra of the Ce 3d, Co 2p, O 1s, Zr 3d, Sn 3d and Ti 2p are shown in Fig. 8. Ce 3d core level spectra (Fig. 8a) for all the samples were deconvoluted into eight contributions. The spin–orbit splitting of Ce 3d5/2 and Ce 3d3/2 was 18.5 eV for all the samples, which was in good agreement with literature.48 The u′′′ satellite peak at about 916.5 eV was the fingerprint of Ce4+ state, and its high intensity and area suggested that the main part of the ceria was in Ce4+ oxidation state. After deconvolution the appearance of bands labeled u′ and v′ were typical for Ce3+ ions, which suggested that both oxidation states Ce4+ and Ce3+ coexist in the surfaces of samples.48 The enhancement of unhomogeneity of the Ce and M (M = Zr, Sn and Ti) atoms could improve the valence change of the Ce (Ce4+ → Ce3+).6 Another reason was probably due to the substitution of Ce4+ by M (M = Zr4+, Sn4+ and Ti4+) ions partly in the calcination process. The spontaneous transformation of Ce4+ (rCe4+ = 0.97 Å) ions to the larger Ce3+ (rCe3+ = 1.10 Å) could compensate for this lattice contraction. The relative intensities of u′ to v′ to the whole eight bands were 12.88%, 20.35%, 23.58% and 30.42%, referring to Ce–Co, Ce–Zr–Co, Ce–Sn–Co and Ce–Ti–Co, respectively, which was shown in Table 3. Obviously, the surface ratios of Ce3+ to (Ce3+ + Ce4+) on the catalysts were higher after introduction M ions, indicating the introduction of M could cause charge imbalance and forming the Ce–M solid solution. It was obvious that the binding energy of Ce shifted to the high field after introduction of other cations. The reason could be explained by the density of electron cloud decreased as the addition of strong electrophilic M cations (Electronegativity: Zr = 1.33, Sn = 1.96 and Ti = 1.54) than Ce (1.10).| Sample | Atomic ratio (%) | ||||||
|---|---|---|---|---|---|---|---|
| XRF (bulk) | XPS (surface) | ||||||
| Ce/M | Co/(Ce + M + Co) | Ce/M | Co/(Ce + M + Co) | Co3+/(Co3+ + Co2+) | Ce3+/(Ce3+ + Ce4+) | Oβ/(Oα + Oβ) | |
| Ce–Co | — | 15.08 (15.00) | — | 10.96 | 61.41 | 12.88 | 27.21 |
| Ce–Zr–Co | 4.08 (4.00) | 14.98 (15.00) | 4.42 | 16.99 | 55.20 | 20.35 | 47.26 |
| Ce–Sn–Co | 4.02 (4.00) | 15.03 (15.00) | 4.62 | 26.28 | 53.35 | 23.58 | 70.09 |
| Ce–Ti–Co | 4.01 (4.00) | 15.01 (15.00) | 4.51 | 15.33 | 49.28 | 30.42 | 71.12 |
 | ||
| Fig. 8 XPS spectra of (Ce 3d, Co 2p, O 1s, Zr 3d, Sn 3d and Ti 2p) of the catalysts. | ||
The Co 2p spectra are displayed in Fig. 8b. A set of Co 2p peaks located at ca. 780 eV and 795 eV corresponding to Co 2p3/2 and Co 2p1/2 respectively. Each peak was the summation of signals from Co2+ and Co3+. In Co3O4, Co2+ has higher binding energy than Co3+.49 From the deconvoluted spectra in Fig. 8b, it was concluded that the peaks at 779 eV and 794 eV with a small satellite signal around 788 eV correspond to Co3+. The peaks at 781 eV and 797 eV were characteristic of Co2+.50 Detailed calculation from the deconvoluted peaks reveals that the intensity ratio of Co3+ and Co2+ was different in different catalysts. The surface atomic ratio of (Co3+)xps to (Co3+ + Co2+)xps was in following order Ce–Co > Ce–Zr–Co > Ce–Sn–Co > Ce–Ti–Co (Table 3). Since this sequence was consistent with the NO conversion performance, the Co3+ on the surface therefore acted as the active sites and its quantity determined the capability of catalytic NO conversion into NO2. Combined with H2-TPR results, we could concluded that Co3+ was more favorable to form in the highly dispersed Co3O4 species.
O 1s spectra were mainly composed of two components. The peak of O 1s named as Oα, with a binding energy of 528–530 eV, contributed to the lattice oxygen in the metal oxides.36 The high binding 531–532 eV energy was attributed to the surface chemisorbed oxygen (Oβ).36 We could find that the O 1s ionization features for all of the catalysts are very similar (Fig. 8c). Obviously, the ratios of chemisorbed oxygen to the whole type of oxygen were higher compared to Ce–Co (Table 3), which indicated that the doping of other cations was beneficial for the formation of chemisorbed oxygen.
In addition, the narrow spectra of Zr 3d, Sn 3d and Ti 2p are shown in Fig. 8d. The binding energy of Zr 3d5/2 (181.8 eV) in the Ce–Zr–Co was consistent with ZrO2 (181.8 eV), indicating that Zr was mainly in a +4 oxidation state.51 As for the XPS spectra of Sn 3d, the most of Sn was mainly in a +4 oxidation state because the binding energy of Sn 3d5/2 (486.2 eV) in Ce–Sn–Co was uniform in SnO2 (486.2 eV).52 For Ce–Ti–Co, the binding energy of Ti 2p3/2 was 458.0 eV, which was lower that in TiO2 (458.3 eV) but higher that in TiO (454.8 eV), indicating that Ti was in a +4 oxidation state and a small quantity on a +3 oxidation state.53
3.6. Relationship between catalytic performance and catalysts properties
It was interesting that different supports could lead to different catalytic performance. In order to understand this question clearly, we attempted to correlate the activity with the above-mentioned characterizations, and the relationship between NO conversion (at 300 °C) and textural/structural/reducible properties was plotted in Fig. 9. As far as we know, for some important catalytic reactions, the increase of the specific surface area of support was beneficial to the dispersion of active species, which could lead the activity to increase; the activity enhanced with decreasing the grain size of catalysts. However, it could be noted from Fig. 9a, the activity couldn't relate to the BET surface area and the grain size very well, we took the dispersion states of Co3O4 and the oxygen vacancies into account. In our previous work,35 we demonstrated that oxygen vacancies were important to the activation of oxygen. Oxygen turned into superoxide ions on oxygen vacancies, superoxide ions could oxide NO for its' strong oxidation ability.54 Furthermore, the dispersion states of the oxides was also a vital factor, Luo et al.13 found the finely dispersed CuO species had the highest catalytic activity among the finely dispersed CuO, the bulk CuO and the Cu2+ in the CeO2 lattice. Highly dispersed Co3O4 species provided more NO adsorption sites and oxygen vacancy were responsible for O2 activation. The NO-TPD profiles are shown in Fig. S1.† The results demonstrated the highly dispersed Co3O4 species were favorable for the NO adsorption. In H2-TPR results, the peak β was ascribed to the reduction of Co3+ to Co2+, the lower reduction temperature may lead to increase the activity. Interestingly, these properties are well in agreement with the activity (Fig. 9b).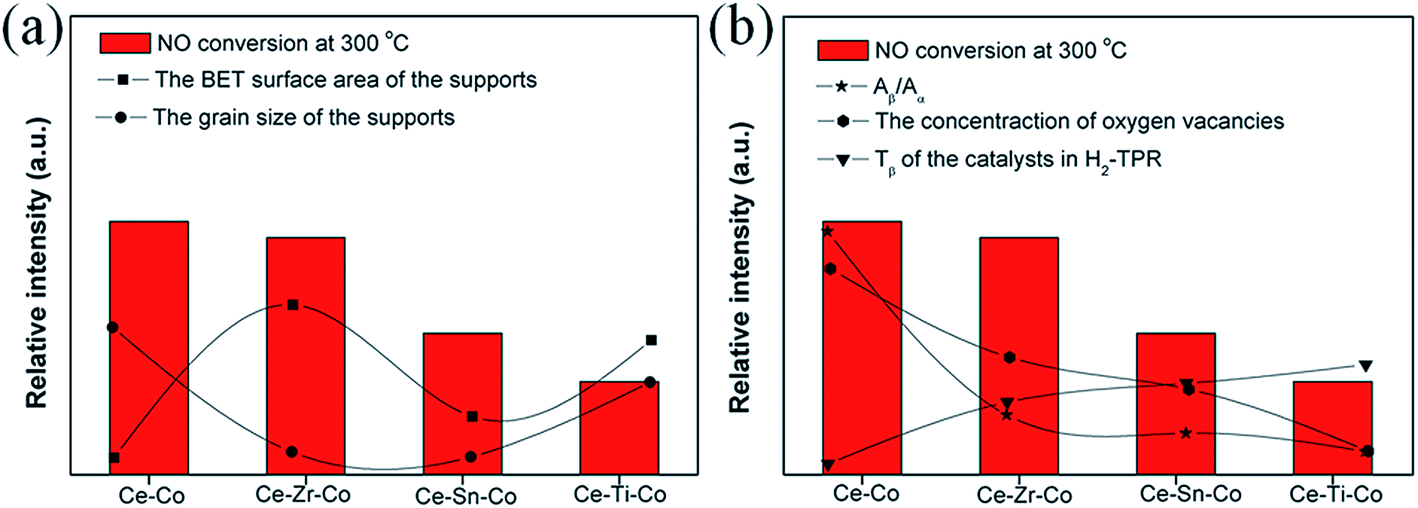 | ||
| Fig. 9 The relationship between the catalytic activity of NO oxidation (at 300 °C) and (a) textural and (b) structural/reducible properties. | ||
4. Conclusion
This work focused on different doped metal cations on the structure, texture, reduction, dispersion states of cobalt species, oxygen vacancies, adsorption properties and catalytic performance of the Co3O4/Ce0.8M0.2O2 (M = Zr4+, Sn4+ and Ti4+) samples for NO oxidation. The catalytic performance results indicated that Co was the active sites. Combined with the above analysis, the catalytic activity of Ce–Co oxides was due to the higher amount of highly dispersed Co3O4, the larger amount of oxygen vacancies and the excellent redox ability.Acknowledgements
This work was financially supported by the Assembly Foundation of The Industry and Information Ministry of the People's Republic of China 2012 (543), the National Natural Science Foundation of China (51408309), Science and Technology Support Program of Jiangsu Province (BE2014713), Natural Science Foundation of Jiangsu Province (BK20140777), Scientific Research Project of Environmental Protection Department of Jiangsu Province (2013003), Industry-Academia Cooperation Innovation Fund Projects of Jiangsu Province (BY2014004-10), Science and technology project of Nanjing (201306012).References
- Z. B. Wu, R. B. Jin, Y. Liu and H. Q. Wang, Catal. Commun., 2008, 9, 2217 CrossRef CAS PubMed.
- G. Madia, M. Koebel, M. Elsener and A. Wokaun, Ind. Eng. Chem. Res., 2002, 41, 3512 CrossRef CAS.
- W. S. Epling, L. E. Campbell, A. Yezerets, N. W. Currier and J. E. Parks II, Catal. Rev.: Sci. Eng., 2004, 46, 163 Search PubMed.
- B. J. Cooper, H. J. Jung and J. E. Thoss, US Patent 4, 902, 487, 1990.
- H. Takeuchi, M. Ando and N. Kizawa, Ind. Eng. Chem. Process Des. Dev., 1977, 16, 303 CAS.
- W. Cai, Q. Zhong, W. Zhao and Y. F. Bu, Appl. Catal., B, 2014, 158–159, 258 CrossRef CAS PubMed.
- J. Giménez-Maňogil, A. Bueno-López and A. García-García, Appl. Catal., B, 2014, 152–153, 99 CrossRef PubMed.
- I. Atribak, A. Bueno-López, A. García-García and B. Azambre, Phys. Chem. Chem. Phys., 2010, 12, 13770 RSC.
- S. K. Megarajana, S. Rayalu, Y. Teraoka and N. Labhsetwar, J. Mol. Catal. A: Chem., 2014, 385, 112 CrossRef PubMed.
- Z. Ren, Y. B. Guo, Z. H. Zhang, C. H. Liu and P. X. Gao, J. Mater. Chem. A, 2013, 1, 9897 CAS.
- M. Sanchez-Dominguez, L. F. Liotta, G. D. Carlo, G. Pantaleo, A. M. Venezia, C. Solans and M. Boutonnet, Catal. Today, 2010, 158, 35 CrossRef CAS PubMed.
- L. Qi, C. J. Tang, L. Zhang, X. J. Yao, Y. Cao, L. F. Liu, F. Gao, L. Dong and Y. Chen, Appl. Catal., B, 2012, 127, 234 CrossRef CAS PubMed.
- M. F. Luo, Y. P. Song, J. Q. Lu, X. Y. Wang and Z. Y. Pu, J. Phys. Chem. C, 2007, 111, 12686 CAS.
- L. Meng, J. J. Lin, Z. Y. Pu, L. F. Luo, A. P. Jia, W. X. Huang, M. F. Luo and J. Q. Lu, Appl. Catal., B, 2012, 119–120, 117 CrossRef CAS PubMed.
- J. Z. Shyu, W. H. Weber and H. S. Gandhi, J. Phys. Chem., 1988, 2, 4964 CrossRef.
- M. Yan, T. Mori, J. Zou, F. Ye, D. R. Ou and J. Drennan, Acta Mater., 2009, 57, 722 CrossRef CAS PubMed.
- F. Chen, J. J. Zhu, Y. Y. Zhan, X. Y. Lin, G. H. Cai, K. M. Wei and Q. Zheng, Appl. Catal., A, 2009, 363, 208 CrossRef PubMed.
- T. Yuzhakova, V. Rakić, C. Guimon and A. Auroux, Chem. Mater., 2007, 19, 2970 CrossRef CAS.
- B. Bonnetot, V. Rakic, T. Yuzhakova, C. Guimon and A. Auroux, Chem. Mater., 2008, 20, 1585 CrossRef CAS.
- S. Somacescu, P. Osiceanu, J. M. C. Morenoa, L. Navarrete and J. M. Serra, Microporous Mesoporous Mater., 2013, 179, 78 CrossRef CAS PubMed.
- M. F. Luo, J. Chen, L. S. Chen, J. Q. Lu, Z. C. Feng and C. Li, Chem. Mater., 2001, 13, 197 CrossRef CAS.
- T. Y. Li, S. J. Chiang, B. J. Liaw and Y. Z. Chen, Appl. Catal., B, 2011, 103, 143 CrossRef CAS PubMed.
- W. T. Chen, K. B. Chen, M. F. Wang, S. F. Weng, C. S. Lee and M. C. Lin, Chem. Commun., 2010, 46, 3286 RSC.
- W. Cai, Q. Zhong, S. L. Zhang and W. Zhao, Chem. Eng. J., 2014, 36, 223 CrossRef PubMed.
- I. Dobrosz-Gómez, I. Kocemba and J. M. Rynkowski, Appl. Catal., B, 2008, 83, 240 CrossRef PubMed.
- J. L. Ayastuy, A. Iglesias-González and M. A. Gutiérrez-Ortiz, Chem. Eng. J., 2014, 244, 372 CrossRef CAS PubMed.
- Y. Wen, C. Zhang, H. He, Y. Yu and Y. Teraoka, Catal. Today, 2007, 126, 400 CrossRef CAS PubMed.
- M. F. Irfan, J. H. Goo and S. D. Kim, Appl. Catal., B, 2008, 78, 267 CrossRef CAS PubMed.
- D. S. Kim, Y. H. Kim, J. E. Yie and E. D. Park, Korean J. Chem. Eng., 2010, 27, 49 CrossRef CAS.
- L. J. Liu, Z. J. Yao, B. Liu and L. Dong, J. Catal., 2010, 275, 45 CrossRef CAS PubMed.
- A. Martínez-Arias, M. Fernández-García, L. N. Salamanca, R. X. Valenzuela, J. C. Conesa and J. Soria, J. Phys. Chem. B, 2000, 104, 4038 CrossRef.
- Q. Q. Jiang, G. L. Zhou, Z. X. Jiang and C. Li, Sol. Energy, 2014, 99, 55 CrossRef CAS PubMed.
- B. Bonnetot, V. Rakic, T. Yuzhakova, C. Guimon and A. Auroux, Chem. Mater., 2008, 20, 1585 CrossRef CAS.
- J. R. McBride, K. C. Hass, B. D. Poindexter and W. H. Weber, J. Appl. Phys., 1994, 76, 2435 CrossRef CAS PubMed.
- S. L. Zhang, H. Y. Li and Q. Zhong, Appl. Catal., A, 2012, 435–436, 156 CrossRef CAS PubMed.
- R. Si, Y. W. Zhang, S. J. Li, B. X. Lin and C. H. Yan, J. Phys. Chem. B, 2004, 108, 12481 CrossRef CAS.
- B. M. Reddy, P. Bharali and P. Saikia, J. Phys. Chem. C, 2008, 112, 11729 CAS.
- Á. Kukovecz, Z. Kónya, D. Mönter, W. Reschetilowski and I. Kiricsi, J. Mol. Struct., 2001, 563–564, 403 CrossRef.
- L. Poul, N. Jouini and F. Fiévet, Chem. Mater., 2000, 12, 3123 CrossRef CAS.
- L. J. Liu, Y. Cao, W. J. Sun, Z. J. Yao, B. Liu, F. Gao and L. Dong, Catal. Today, 2011, 175, 48 CrossRef CAS PubMed.
- J. Y. Luo, M. Meng, X. Li, X. G. Li, Y. Q. Zha, T. D. Hu, Y. N. Xie and J. Zhang, J. Catal., 2008, 254, 310 CrossRef CAS PubMed.
- P. Arnoldy and J. A. Moulijn, J. Catal., 1985, 93, 38 CrossRef CAS.
- M. Kang, M. W. Song and C. H. Lee, Appl. Catal., A, 2003, 251, 143 CrossRef CAS.
- C. W. Tang, C. C. Kuo, M. C. Kuo, C. B. Wang and S. H. Chien, Appl. Catal., A, 2006, 309, 37 CrossRef CAS PubMed.
- L. F. Liotta, G. Di Carlo, G. Pantaleo, A. M. Venezia and G. Deganello, Appl. Catal., B, 2006, 66, 217 CrossRef CAS PubMed.
- L. Spadaro, F. Arena, M. L. Granados, M. Ojeda, J. L. G. Fierro and F. Frusteri, J. Catal., 2005, 234, 451 CrossRef CAS PubMed.
- A. Martínez-Arias, M. Fernández-García, O. Gálvez, J. M. Coronado, J. A. Anderson, J. C. Conesa, J. Soria and G. Munuera, J. Catal., 2000, 195, 207 CrossRef.
- E. Béche, P. Charvin, D. peranau, S. Abanades and G. Flamant, Surf. Interface Anal., 2008, 40, 264 CrossRef.
- M. Yung, E. Holmgreen and U. Ozkan, Catal. Lett., 2007, 118, 180 CrossRef CAS.
- Y. Ding, L. Zhu, A. Huang, X. Zhao, X. Zhang and H. Tang, Catal. Sci. Technol., 2012, 2, 1977 CAS.
- S. Velu, K. Suzuki, C. S. Gopinath, H. Yoshida and T. Hattori, Phys. Chem. Chem. Phys., 2002, 4, 1990 RSC.
- D. Shuttleworth, J. Phys. Chem., 1980, 84, 1629 CrossRef CAS.
- B. M. Reddy, P. Lakshmanan and A. Khan, J. Phys. Chem. B, 2004, 108, 16855 CrossRef CAS.
- Z. Zhao, X. G. Yang and Y. Wu, Appl. Catal., B, 1996, 8, 281 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra15439h |
| This journal is © The Royal Society of Chemistry 2015 |