
Multi-omics based changes in response to cadmium toxicity in Bacillus licheniformis A
Jing Sunab,
Jun Zhoua,
Zhonghua Wanga,
Weina Hea,
Dijun Zhanga,
Qianqian Tonga and
Xiurong Su†
*a
aSchool of Marine Sciences, Ningbo University, Ningbo 315211, China. E-mail: suxiurong@nbu.edu.cn; Fax: +86-574-87608368; Tel: +86-574-87608368
bCollege of Food Science and Technology, Nanjing Agricultural University, Nanjing 210095, China
First published on 19th December 2014
Abstract
Cadmium (Cd), a widespread substance with high toxicity and persistence, is known to cause a broad range of adverse effects in all living organisms. However, many microorganisms have special detoxification mechanisms for Cd. In this study, we investigated the response to Cd in Bacillus licheniformis A at the omics level. Proteomic biomarkers were identified, including peroxiredoxin, nucleoside diphosphate kinase, trigger factor, and elongation factor Tu. These proteins suggested that Cd could induce oxidative stress, disturbance in energy metabolism, and protein synthesis in B. licheniformis A. Furthermore, metabolites (glutamate, glycine, and S-adenosylhomocysteine) involved in the glutathione synthesis pathway were also affected. In addition, some metabolic biomarkers were validated by relating to respective proteins, including 50S ribosomal protein L7/L12, trigger factor, peroxiredoxin and superoxide dismutase in B. licheniformis A. This study provides new insights into the utilization of an omics approach in bacteria to assess the effects of Cd pollution in the environment.
1. Introduction
Environmental pollution caused by cadmium (Cd) has received much attention in recent years because of its toxicity and stability. Sources of Cd include metallurgy, mining, fossil fuel combustion, the manufacture and disposal of rechargeable nickel–cadmium batteries, and the utilization of pesticides and fertilizers.1,2 It binds to organic compounds by forming bonds with nitrogen and sulfur, inactivating proteins, and potentially leading to negative side-effects, including extreme toxicity, carcinogenicity, and mutagenicity.3 It is easily absorbed and accumulated by lower organisms and subsequently transferred to higher trophic levels in food chains. Due to Cd's severe toxic effects, fast accumulation through the food chains has caused serious damage to ecosystems and led to economic losses and negative impacts on food chains and human health. For this reason, the International Agency for Research on Cancer has classified Cd as a human carcinogen belonging to group 1 agents in 1993.4Bioremediation with microorganisms has become an attractive alternative to conventional techniques for toxin removal. Furthermore, there are some studies that have demonstrated the efficiency of metal removal by bacteria.5–7 Some microorganisms can tolerate heavy metals by using strategies such as exclusion by permeability barriers, intra- and extra-cellular sequestration, resistance to oxidative stress, extracellular biosorption, active transport efflux pumps, enzymatic detoxification, and a reduction in the sensitivity of cellular targets to metal ions.8–11 For example, as a Cd2+-resistance determinant the CadA gene in Listeria monocytogenes encodes an integral protein which belongs to the P-type ATPase family.12 These ATPases are membrane proteins coupling ATP hydrolysis and active transport of cations or anions across the membrane in which they are embedded.
Toxicity of various heavy metals to bacteria in the environment has been extensively studied for many years.13–17 Recently, omics technologies have increasingly been used in a variety of different contexts to gain a comprehensive understanding of nearly all components within a biological entity. They include transcriptomics, proteomics and metabolomics, which refer to the pool of RNA transcripts, proteins and metabolites in a cell, tissue biofluid, or indeed whole organisms.3,18 These technologies and related data processing techniques provide powerful tools which have the potential to be of great value in ecotoxicological research.18–23 Proteomics has been widely used to study the mechanisms of heavy metal toxicity in bacteria. Mass spectrometry (MS), two-dimensional electrophoresis (2-DE), protein affinity techniques, high-throughput immunochemistry and bioinformatics are among the major proteomics technologies used for understanding metal toxicity. Kılıç et al. used a proteomics approach to study the Cr(VI) stress response of Pseudomonas aeruginosa.9 Additionally, Siripornadulsil et al. identified and characterized protein expression in Cupriavidus taiwanensis KKU2500-3 during growth under Cd stress by using 2-DE and MS.24 Metabolites, as the end products of cellular regulatory processes, can be regarded as the ultimate response of biological systems to genetic or environmental changes.3 Metabolomic analyses have multiple advantages in that results obtained at a molecular biological level may also provide insight into mechanisms of toxicity.25
Bacillus licheniformis, a Gram-positive soil bacterium, classified as GRAS (generally recognized as safe), was shown to be an efficient plant growth promoting rhizobacterium.26 Earlier study showed B. licheniformis is able to improve the growth and development of the host plant in Cd contaminated soils.27 However, little information is available regarding the mechanisms of B. licheniformis resistance to Cd.
In this study, proteomic, transcriptomic and metabolomic analyses were combined to explore the effects of Cd exposure in B. licheniformis A. Bacterial proteins were isolated by 2-DE and identified by peptide mass fingerprinting (PMF). Differential gene transcription of some annotated candidates was further assessed by Real Time PCR while proton (1H) nuclear magnetic resonance (NMR) spectroscopy was utilized for the metabolomic analyses.
2. Materials and methods
2.1. Bacterial strain
B. licheniformis A (CGMCC no. 4324) is isolated and characterized in our laboratory. A single colony was propagated in beef extract peptone medium at 37 °C overnight. The cultures were separately used to determine the minimum inhibitory concentrations (MICs) for an ancestor strain by Cd and for resistant strains by progressive Cd2+ gradient concentration domestication. Growth was monitored spectrophotometrically (measurement of the optical density (OD)) at 560 nm at intervals of 6 h for 48 h.2.2. Transmission electron microscopy analysis
Transmission electron microscopy (TEM) analysis of the bacteria before and after coming in contact with Cd2+ was carried out as follows: firstly, the cell pellets were fixed using 2.5% (v/v) of glutaraldehyde in 0.1 M phosphate buffered saline (PBS, pH 7.4) for 2–3 h at 4 °C followed by washing with 0.1 M PBS, post-fixation was carried out with 2% (v/v) osmium tetroxide for 2 h in the same buffer at 4 °C. After several washes in buffer, the cells were dehydrated using increasing concentrations of 99.9% (v/v) ethanol (30, 50, 70, 80, 90, and 100%) at 10 min intervals followed by 5 min washing in 100% (v/v) acetone. The cells were then embedded in 50% and 100% (v/v) of epoxy resin in acetone for 15 min each. The cell pellets were again infiltrated with 100% (v/v) epoxy resin and cured at 60 °C overnight. Specimens of 100 nm thickness were sectioned from the embedded blocks using a microtome (HM505E, Carl Zeiss International, Germany) and mounted on 200-mesh copper grids. The specimens were stained with uranyl acetate, post-stained with lead-citrate and viewed using a transmission electron microscope (TEM) (H-7650, Hitachi Ltd., Japan).2.3. Proteomic analysis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) and incubated at −20 °C overnight. The protein pellet was further centrifuged at 12000g for 30 min at 4 °C and resuspended in 7 M urea, 2 M thiourea, 4% CHAPS, 65 mM dithiothreitol (DTT), and 0.2% Bio-Lyte (Bio-Rad) sample buffer (for isoelectric focusing (IEF)). Protein concentrations were determined using a non-interference protein assay kit (Sangon Biotech Co., Ltd, Shanghai, China) against a bovine serum albumin standard curve. Protein samples were stored at −80 °C for further analysis.000 V h and a maximum voltage of 4000 V. After the strip was equalized in 2% (w/v) DTT and 2.5% (w/v) iodoacetamide (IAA) (6 M urea, 20% glycerol, 2% SDS, 0.375 M pH 8.8 Tris-HCl) for 15 min, respectively, proteins were separated in the second dimension by using a 12% acryl amide SDS-PAGE gel and electrophoresed with voltages of 80 V and 120 V. All 2-DE experiments were carried out in triplicate.
:4) and incubated at −20 °C overnight. The protein pellet was further centrifuged at 12000g for 30 min at 4 °C and resuspended in 7 M urea, 2 M thiourea, 4% CHAPS, 65 mM dithiothreitol (DTT), and 0.2% Bio-Lyte (Bio-Rad) sample buffer (for isoelectric focusing (IEF)). Protein concentrations were determined using a non-interference protein assay kit (Sangon Biotech Co., Ltd, Shanghai, China) against a bovine serum albumin standard curve. Protein samples were stored at −80 °C for further analysis.000 V h and a maximum voltage of 4000 V. After the strip was equalized in 2% (w/v) DTT and 2.5% (w/v) iodoacetamide (IAA) (6 M urea, 20% glycerol, 2% SDS, 0.375 M pH 8.8 Tris-HCl) for 15 min, respectively, proteins were separated in the second dimension by using a 12% acryl amide SDS-PAGE gel and electrophoresed with voltages of 80 V and 120 V. All 2-DE experiments were carried out in triplicate.The samples were analyzed on an Autoflex™ speed matrix-assisted laser desorption ionization time-of-flight mass spectrometer (Bruker Daltonics, Bremen, Germany) using a 200 Hz, 355 nm nitrogen laser for desorption and ionization. Default mode was used with an acquisition mass range from 700 to 3200 Da and the total acceleration voltage was 20 kV. The instrument was internally calibrated using tryptic autolytic peaks. All spectra were smoothed, noise filtered, and peak lists generated using flexAnalysis version 2.0 (Bruker). Then, the measured tryptic peptide masses were transferred to the BioTools (Bruker) program to be searched against the taxonomy of Bacteria in the protein database using the Mascot search engine (http://www.matrixscience.com). For protein searches, monoisotopic masses were used, considering a peptide tolerance of 50 ppm and allowance of one missed tryptic cleavage site. When MS/MS was carried out, a tolerance of 0.5 Da was acceptable. Fixed modifications were set for carbamidomethylation of cysteine and variable modifications were set for oxidation of methionine. Protein identifications were considered as reliable when the Mascot score was ≥84 (Mascot score was calculated as −10 × logP, where P is the probability that the observed match is a random event). This is the lowest score indicated by the program as significant (p < 0.05) and indicated by the probability of incorrect protein identification. For searches done with identified proteins were only considered if having at least two peptides with 95% confidence. They were further validated with inspection of the assigned sequence. Functional characterization of the proteins was predicted with Expert Protein Analysis System (Expasy) proteomics server of the Swiss Institute of Bioinformatics (SIB) (http://www.expasy.org/) based on Gene Ontology (GO) annotation under the category of biological process in UniProtKB (http://www.uniprot.org).
2.4. Confirmation of differentially expressed proteins by real time PCR
Real time PCR (RT-PCR) was performed using three replicates for different concentrations of Cd treatment groups. Total RNA was extracted from log-phase B. licheniformis A with Trizol reagent (Takara), according to the manufacturer's protocol. Single-strand cDNA synthesis was performed according to the M-MLV cDNA Reverse Transcription Kit (Sangon) Usage information. The reactions were incubated at 42 °C for 1 h, and terminated by heating at 70 °C for 10 min. RT-PCR was performed for 4 genes differentially expressed during Cd treatment using gene specific primers (see Table 1). Primers were chosen to have melting temperatures of 59–60 °C and the amplified product size was approximately 200 base pairs long. 16s rRNA of B. licheniformis A was used as a reference housekeeping gene for normalization. The RT-PCR amplifications were carried out in a total volume of 20 μL containing 10 μL 2 × SYBR Green Mix (Takara), 4 μL of the 1:20 diluted cDNA, 1 μL of each primer (20 mM) and 4 μL PCR grade water. RT-PCR amplification was performed using a Rotor-Gene 6000 real-time PCR detection system (Qiagen, Germany). The RT-PCR parameters were as follows: denaturation at 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s, 60 °C for 20 s, 72 °C for 20 s. Melting analysis of the amplified products was performed at the end of each PCR run to confirm that a single PCR product was produced and detected. Quantification of the PCR product was performed using the −△△Ct method. The value obtained denoted the n-fold difference relative to the calibrant (control samples). The data were presented as relative mRNA expression levels (means ± S.D., n = 3), and the results subjected to paired t-test analysis and p ≤ 0.05 were considered statistically significant.
| Primer | Sequence (5′–3′) | Name |
|---|---|---|
| P1 | F-CGGCGTGCCTAATACATGC | 16s rRNA (reference gene) |
| P2 | R-GGCAGGTTACCCACGTGT | |
| P3 | F-ACGGTGTTGCAATCGATCCT | PTS system glucose-specific transporter subunit |
| P4 | R-CGTGAGCTTCGAAACCTTCAC | |
| P5 | F-GAAGACGCTCAAAAACGCGT | Trigger factor |
| P6 | R-CGCTTCCAAGAGCTTGCTTG | |
| P7 | F-ATTCGGTAGCTTCGATGCGT | Superoxide dismutase |
| P8 | R-CGTTTTTCAGCAGCGTTCCA | |
| P9 | F-CGCTTGAAGCTGGCCTTG | Universal stress protein |
| P10 | R-CAAGGACATCGCATTTCGCA |
2.5. Metabolomics analysis
:1, v/v) and sonicated for 15 min. Cell debris was removed by centrifugation at 8000 g for 10 min. The supernatants were evaporated to dryness and stored at −80 °C prior to NMR analysis.000 rpm for 30 min at 4 °C. 450 μL of the supernatant and 50 μL 5 mM Anachro Certified DSS Standard Solution were mixed together then transferred into 5 mm NMR tube. The samples were analyzed using a Bruker AV III 600 MHz spectrometer equipped with an inverse cryoprobe operating at 600.13 MHz for 1H frequency at 298 K. Spectra were collected using a solvent suppression pulse sequence based on a 1-dimensional NOESY sequence with presaturation during relaxation delay and mixing time. Generally, 32 free induction decays (FIDs) and 128 transients were collected for each sample over a spectral width of 12019.231 Hz at 25 °C according to Jamers et al.33. Results
3.1. Effect of Cd2+ on the growth of B. licheniformis A strain and TEM analysis
To study the effect of Cd2+ on the growth of B. licheniformis A, the cells were cultivated in the presence of increasing concentrations of Cd2+. The measured optical densities (at 560 nm) of control and Cd2+ treated cells over time are shown in Fig. 1(a). Growth rate were significantly affected by Cd2+ decreasing from 0.258 h−1 in the control sample to less than 0.081 h−1 in the presence of 1000 ppm Cd2+. B. licheniformis A cells treated with 500 ppm Cd2+ did not show obvious differences in growth rate relative to the control sample. However, the lag phase for 1000 ppm Cd2+ contaminated cultures was longer and its maximum growth rate was slower than the control. It can be seen from Fig. 1(a) that the stress-free cells reached the mid-log phase faster than the stressed cells which needed a longer time period for adaptation. Finally, we chose the strains that were cultured in a beef extract peptone medium with 1000 ppm Cd2+ as the experimental group and the samples lacking Cd2+ as the control group for the proteomic studies.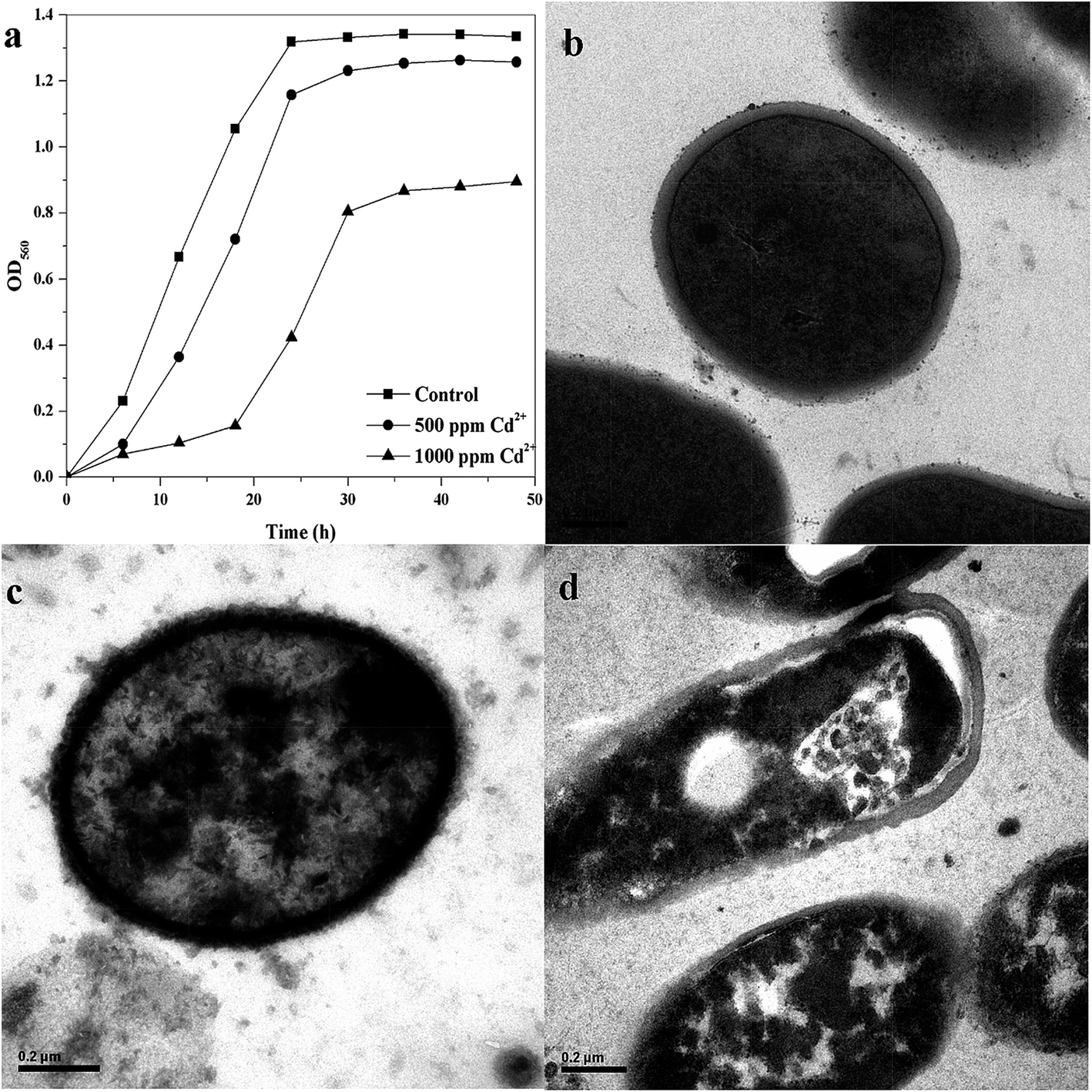 | ||
| Fig. 1 Growth curve of B. licheniformis A cultured without or with different concentrations of Cd2+ (a), and related control (b), 500 ppm Cd2+ (c), and 1000 ppm Cd2+ (d) TEM micrographs. | ||
Transmission electron micrographs of varying concentrations of Cd2+-treatment cells showed that the surface of the normal cell is smooth, with even edges (Fig. 1(b)). However, while the bacterial cell structure remained intact with the increase of Cd2+ concentration (Fig. 1(c) and (d)), separation between the bacterial cell wall and membrane was noticeable (Fig. 1(d)). A noticeable strongly electron-dense area between the cell wall and cytoplasm clearly indicates the deposition of Cd2+ onto the bacterial cells functional groups with further translocation into the cytoplasmic region (Fig. 1).
3.2. Differential protein expression
A comparative proteomic analysis was used to detect significantly up- or down-regulated proteins in B. licheniformis A in response to the presence of 1000 ppm Cd2+ stress. An average of 245 protein spots were detected in the control group gel and 327 protein spots were detected in the experimental group. From these, a total of 145 spots were matched in all gels. A total of 18 spots were selected for identification by MS/MS (Fig. 2). Of these, 13 proteins were up-regulated and 5 proteins down-regulated upon Cd2+ stress (Table 2). These proteins were found to be related to energy and primary metabolic processes (PTS system glucose-specific transporter subunit IIA, nucleoside diphosphate kinase, dihydrolipoamide dehydrogenase, acetoin dehydrogenase complex), stress and defense (universal stress protein, trigger factor, peroxiredoxin, superoxide dismutase, manganese superoxide dismutase, hypothetical protein IC5_00055), cell growth/division (naphthoate synthase, putative delta-1-pyrroline-5-carboxylate dehydrogenase, 1-pyrroline-5-carboxylate dehydrogenase, spore coat-associated protein N, response regulator aspartate phosphatase), and signal transduction (elongation factor Tu, 50S ribosomal protein L7/L12). | ||
| Fig. 2 Representative 2-DE images of proteins from B. licheniformis A cultured without (a) or with 1000 ppm Cd2+ (b). The spots in black boxes were selected to be identified by MS/MS and listed in Table 2. | ||
| Spot noa | Accession no. | Protein name | Mr/pIb | Protein scorec | Sequence coverage | Expect | Matched peptides | Fold changed |
|---|---|---|---|---|---|---|---|---|
| a Assigned spot no. as indicated in Fig. 2.b Experimental mass and pI.c Mascot score reported.d Fold changes with significances (>1.5 folds and p < 0.05) were calculated using PDQuest 2DE analysis (Version 8.0.1) software. | ||||||||
| 1 | gi|423370999 | Hypothetical protein IC5_00055 | 87411/5.92 | 444 | 41% | 5.6 × 10−38 | 22 | 3.58 |
| 2 | gi|206974153 | Peroxiredoxin | 20864/4.81 | 425 | 56% | 4.4 × 10−36 | 10 | 2.34 |
| 3 | gi|301055460 | Dihydrolipoamide dehydrogenase | 49395/5.13 | 274 | 44% | 5.6 × 10−21 | 13 | 2.05 |
| 4 | gi|30265343 | PTS system glucose specific transporter subunit IIA | 17464/5.41 | 343 | 73% | 7 × 10−28 | 9 | 2.60 |
| 5 | gi|217961965 | Trigger factor | 47185/4.53 | 283 | 52% | 7 × 10−22 | 19 | New |
| 6 | gi|206977024 | Naphthoate synthase | 29886/5.56 | 403 | 52% | 7 × 10−34 | 14 | 1.69 |
| 7 | gi|30018370 | 50S ribosomal protein L7/L12 | 12510/5.72 | 92 | 21% | 8.1 × 10−3 | 8 | 3.56 |
| 8 | gi|254739504 | Elongation factor Tu | 41570/4.77 | 250 | 55% | 1.4 × 10−18 | 17 | New |
| 9 | gi|30264347 | Superoxide dismutase | 22650/5.30 | 337 | 34% | 2.8 × 10−27 | 5 | 2.06 |
| 10 | gi|306447497 | Manganese superoxide dismutase | 22616/5.33 | 168 | 59% | 2.2 × 10−10 | 8 | 1.53 |
| 11 | gi|52140879 | Universal stress protein | 16842/5.68 | 189 | 84% | 1.8 × 10−12 | 11 | 1.55 |
| 12 | gi|301053152 | Nucleoside diphosphate kinase | 18849/5.26 | 315 | 60% | 4.4 × 10−25 | 11 | 1.59 |
| 13 | gi|206977864 | Spore coat-associated protein N | 21901/5.76 | 292 | 43% | 8.9 × 10−23 | 8 | −2.78 |
| 14 | gi|30265465 | Superoxide dismutase | 24122/5.40 | 156 | 49% | 3.5 × 10−9 | 7 | 1.63 |
| 15 | gi|206973646 | Acetoin dehydrogenase complex | 49619/5.65 | 151 | 25% | 1.1 × 10−8 | 7 | −2.89 |
| 16 | gi|229035348 | Response regulator aspartate phosphatase | 43565/5.73 | 446 | 49% | 3.5 × 10−38 | 17 | −1.52 |
| 17 | gi|196040213 | Putative delta-1-pyrr Oline-5-carboxylate dehydrogenase | 56446/5.43 | 249 | 29% | 1.8 × 10−18 | 12 | −3.86 |
| 18 | gi|229089424 | 1-Pyrroline-5-carboxylate dehydrogenase | 56445/5.51 | 432 | 48% | 8.9 × 10−37 | 20 | −1.92 |
3.3. mRNA-level expression
To further verify the results of protein responses and compare the correlation between protein abundances and gene expressions, four representative proteins, including PTS system glucose specific transporter subunit IIA (spot 4), trigger factor (spot 5), superoxide dismutase (spot 9) and universal stress protein (spot 11), were quantified using qRT-PCR. As shown in Fig. 3, the gene expressions levels of spot 4 (34-fold), spot 9 (26-fold) and spot 11 (5-fold) were positively correlated with the corresponding proteins (2.60-fold, 2.06-fold and 1.55-fold) during Cd2+ treatment. The expression of spot 5 (16-fold), as a new protein compared with control, was also observed on the mRNA level.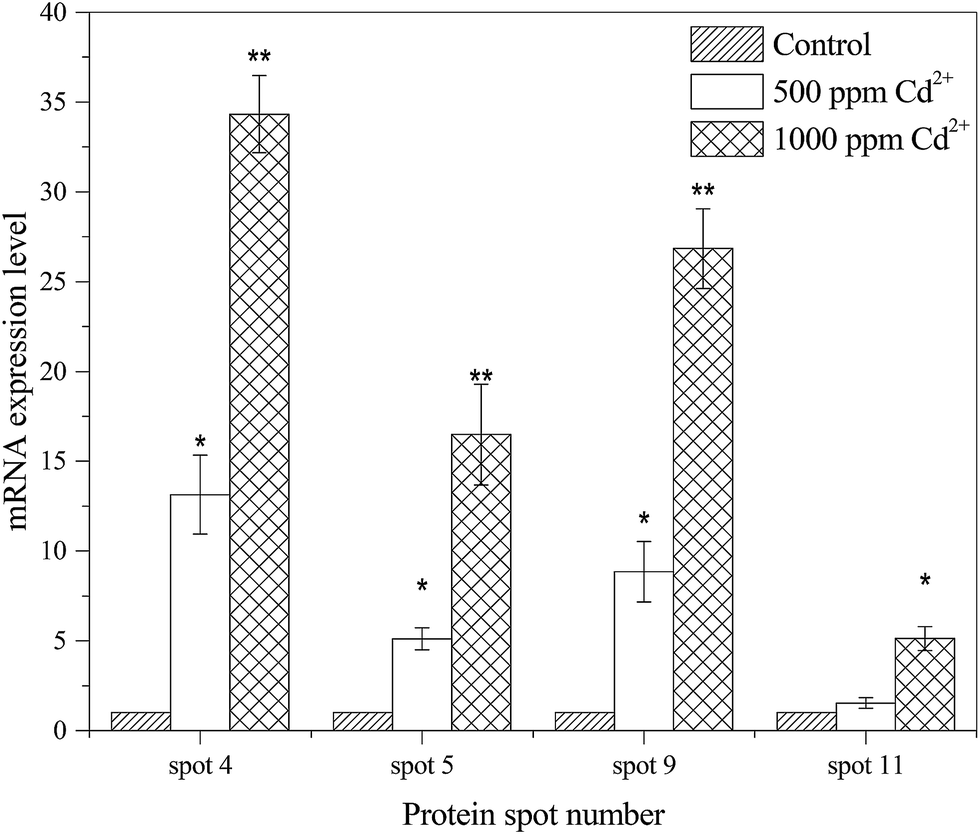 | ||
| Fig. 3 qRT-PCR analysis of gene expression level in different Cd2+ concentrations treated B. licheniformis A. The mRNA expression levels of the ratio of treatment groups to the control are plotted (*P < 0.05, **P < 0.01). | ||
3.4. Metabolite profiling
Fig. 4 shows the representative 1H NMR spectra of B. licheniformis A, including amino acids and derivatives (alanine, asparagine, glutamate, glycine, isoleucine, leucine, methionine, valine, S-adenosylhomocysteine, etc.), ammoniums compounds (choline, O-phosphocholine), nucleic acid components (adenosine, cytidine, guanosine, inosine, oxypurinol, uracil, uridine) and organic acids (acetate, formate, lactate, succinate). | ||
| Fig. 4 Representative 1-dimensional 600 MHz 1HNMR spectra of B. licheniformis A extracts. Keys: (1) isoleucine, (2) leucine, (3) valine, (4) ethanol, (5) lactate, (6) alanine, (7) lysine, (8) acetate, (9) glutamate, (10) methionine, (11) succinate, (12) aspartate, (13) asparagine, (14) phenylalanine, (15) betaine, (16) threonine, (17) uridine, (18) adenosine, (19) uracil, (20) guanosine, (21) cytidine, (22) S-adenosylhomocysteine, (23) inosine, (24) tyrosine, (25) oxypurinol, (26) formate, (27) 4-aminobutyrate, (28) ethylene glycol, (29) glycine. | ||
Principal component analysis (PCA) was performed on the NMR spectral datasets of B. licheniformis A extracts from control and Cd2+-treated groups. Examination of the score plot (Fig. 5) in the area defined by the first two principal components (97.8% of the total variance) revealed two clusters of B. licheniformis A extracts with a clear separation along the first principal component (PC1, 95.9%). The second principal component (PC2) explained 1.9% of the variance. The PCA resulted in separations between control and Cd2+-treated groups, as shown in Fig. 5.
 | ||
| Fig. 5 Principal component analysis (PCA) score plot of PC1 versus PC2 for 1H NMR spectra of B. licheniformis A extracts (n = 5). The score plot in the area defined by the first two principal components (97.8% of the total variance) revealed two clusters according to Cd2+-treatment. | ||
Further analysis was performed on the NMR spectral data and quantified metabolite concentrations to identify potential metabolic biomarkers induced by Cd2+ (Fig. 6). The concentrations of almost all of identified metabolites were significantly decreased in Cd2+-treatment samples, especially in two different sulfur-containing amino acids. In contrast, the concentration of lactate increased upon Cd2+ stress. Compared to these metabolites in control group, choline and O-phosphocholine were uniquely detected in Cd2+-treatment group. In agreement with B. licheniformis from Genes and Genomes (http://www.genome.jp/kegg/) and Uniprot (http://www.uniprot.org/), Fig. 7 displays the schematic presentation of whole proteomic and metabolomic responses in B. licheniformis A challenged by Cd2+.
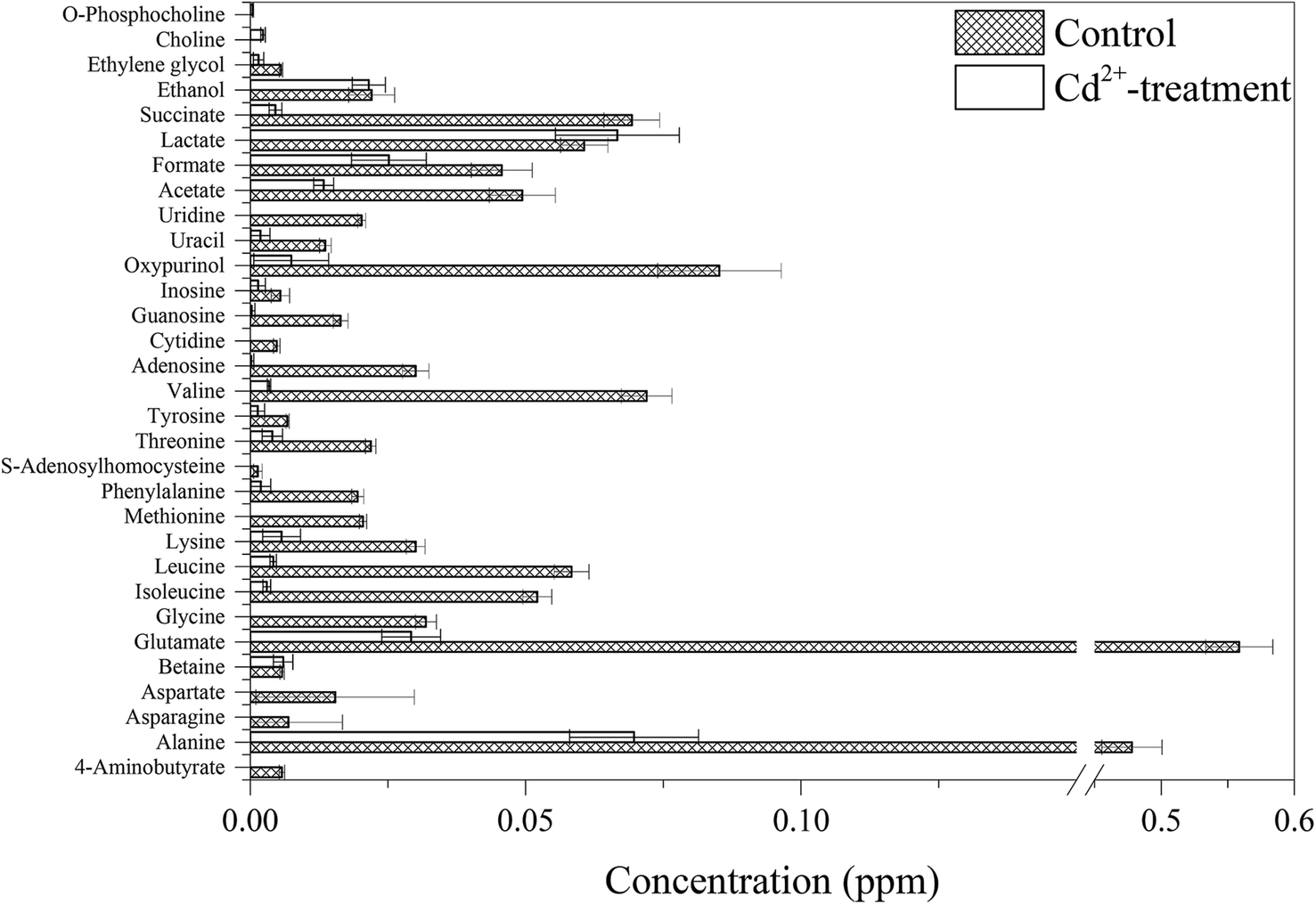 | ||
| Fig. 6 Changes in metabolite concentrations in B. licheniformis A between control and Cd2+-treatment groups. | ||
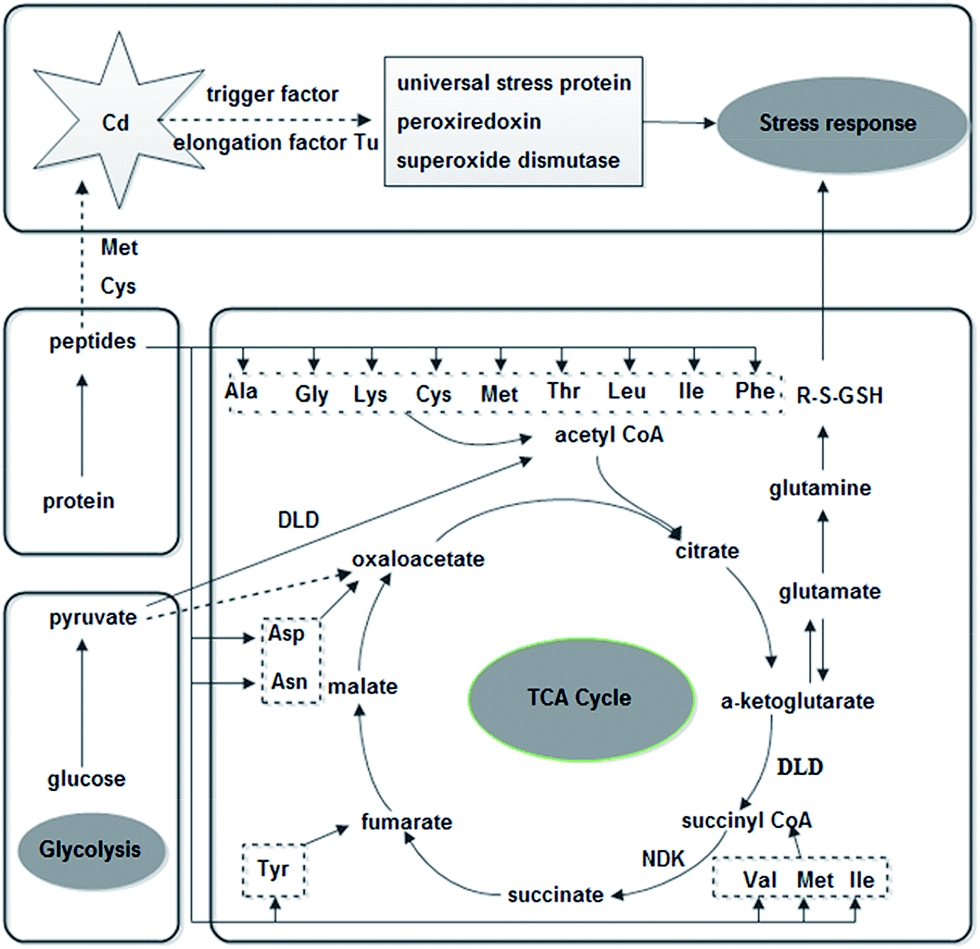 | ||
| Fig. 7 Schematic representation of molecular response mechanisms in B. licheniformis A with Cd2+-treatment according to B. licheniformis of Genes and Genomes (http://www.genome.jp/kegg/) and Uniprot (http://www.uniprot.org/). Abbreviation: ala, Alanine; Asp, aspartate; Asn, aspsrsgine; Cys, S-adenosylhomocysteine; DLD, dihydrolipoamide dehydrogenase; Gly, glycine; GSH, glutathione; Ile, isoleucine; Leu, leucine; Lys, lysine; Met, methionine; NDK, nucleoside diphosphate kinase; NS, naphthoate synthase; Phe, phenylalanine; PTS, PTS system glucose specific transporter subunit IIA; RP, 50S ribosomal protein L7/L12; Thr, threonine; Tyr, tyrosine; Val, valine. | ||
4. Discussion
4.1. Effects of Cd2+ on the proteome of B. licheniformis A
B. licheniformis A modulated the expression of several proteins when exposed to high concentrations of Cd2+. Fig. 7 demonstrated the pathways involved in the response of B. licheniformis A to Cd2+ challenge. In Cd2+-treated samples, the responsive proteins (Table 2) related to metabolism were involved in energy and primary metabolisms. In this study, the significant up-regulation of the PTS system glucose specific transporter subunit IIA might imply that this protein contributes to the defense system of B. licheniformis A. The phosphoenolpyruvate-dependent sugar phosphotransferase system (sugar PTS), which is a major carbohydrate active transport system, catalyzes the phosphorylation of incoming sugar substrates concomitantly with their translocation across the cell membrane.32 Therefore, the up-regulation of PTS system glucose specific transporter subunit IIA probably indicated that bacteria up-regulate their energy transportation system components to meet the needs of their growth under heavy metal stress. Dihydrolipoamide dehydrogenase (DLD) belongs to the class-I pyridine nucleotide-disulfide oxidoreductase family. It serves to re-oxidize lipoic acid residues in the formation of acyl-CoA or succinyl-CoA in the citric acid (TCA) cycle.33 Nucleoside diphosphate kinases (NDKs) are enzymes which catalyze the exchange of phosphate groups between various nucleoside diphosphates.18 In the TCA cycle, NDKs can convert guanosine triphosphate (GTP) to adenosine triphosphate (ATP). Because the bacteria required ATP as an energy source under Cd stress, the NDK was probably up-regulated in B. licheniformis A to produce more ATP to compensate for the energy loss, and stressed growth conditions.Some stress proteins, including molecular chaperones, heat shock proteins and starvation proteins, are easily induced in response to a wide range of stress challenges, such as heavy metals, starvation, oxidation, anaerobiosis, unsuitable temperatures and different pH value.9 Rince et al. demonstrated that the general stress proteins DnaK and GroEL were induced in Enterococcus faecalis in response to heat, ethanol, bile salts, NaCl, H2O2 and pH shifts.34 Under Cd stress, the up-regulation of universal stress protein might imply that this protein contributes to the defense system of B. licheniformis A. Molecular chaperones are wildly distributed stress proteins conserved among almost all organisms. In addition to helping newly synthesized proteins, molecular chaperones play a key role in protein export. Trigger factor which is reported to function as a peptidyl-prolyl cis–trans isomerase, acts as a chaperone by maintaining the newly synthesized protein in an open conformation.35 This protein was solely expressed in the Cd-treatment group, possibly reflecting a series of adaptive changes when faced with environmental stress.
There is a mass of literature showing that Cd can induce oxidative stress in various species.2,3,8,36 The up-regulation of peroxiredoxin, superoxide dismutase, manganese superoxide dismutase was observed in this study suggesting that antioxidant enzymes are useful in protection against Cd toxicity.
Elongation factor Tu (EF-Tu) is a protein which promotes the GTP-dependent binding of aminoacyl-tRNA to the A-site of ribosome during protein biosynthesis.37 The new induction of EF-Tu was also showed in P. fluorescens ATCC 948 treated with 0.1 mM cobalt.38 Ribosomal proteins are also over-expressed at high heavy metal concentrations. The up-regulation of proteins involved in protein biosynthesis is a common response to heavy metals. This result was consistent with the finding by Manara et al.14 EF-Tu and 50S ribosomal protein L7/L12 are intimately involved in protein biosynthesis, reflecting the need for a vigorous synthesis of protective proteins against Cd. Interestingly, Schroeter et al. found many down-regulated proteins involved in translation (e.g. most aminoacyl-tRNA synthetases and some translation elongation factors) after H2O2 treatment in B. licheniformis.39 This difference might be caused by the different oxidative stress between Cd and H2O2.
4.2. Effects of Cd2+ on the metabolome of B. licheniformis A
In Cd2+-treated samples, the concentrations of most identified metabolites were down-regulated (Fig. 6). Amino acids are not only building-blocks in protein synthesis but some are precursors and carbon sources for the TCA cycle (Fig. 7). The consistency between the down-regulation noted in amino acids and proteins including EF-Tu, 50S ribosomal protein L7/L12, trigger factor, peroxiredoxin and superoxide dismutase confirmed the disturbance in protein synthesis. In addition, two kinds of sulfur-containing amino acids may be combined with Cd through thiol groups. Furthermore, a reduction was observed in the levels of glutamate, glycine and S-adenosylhomocysteine, compounds which are necessary for the synthesis of glutathione (GSH). It is a tripeptide, which is synthesized from the amino acids cysteine, glutamate and glycine.3 GSH is the most abundant intracellular thiol in all living cells and is known to be involved in many biological processes, including the synthesis of proteins and DNA, transport, enzyme activity modulation and metabolism as well as defense against reactive oxygen species (ROS).40,41 Bianucci et al. demonstrated that Cd increases GSH concentrations as cells respond by producing increased antioxidant capacity.42In our research, the concentration of lactate was increased. Lactate can be converted to pyruvate. And, pyruvate as an end product of glycolysis can repair the cell damage through antioxidation and modulation of energy metabolism.43
In comparison with the metabolites detected in the control group, choline and O-phosphocholine were uniquely detected in the Cd2+-treated group. However, the Gram-positive model organism Bacillus subtilis are not known to naturally contain phosphocholine as a cellular membrane phospholipids.44 Therefore, choline and O-phosphocholine might be used to resist Cd toxicity.
5. Conclusions
In summary, we have studied the effects of Cd on B. licheniformis A adopting an omic approach. The results showed that Cd could induce the synthesis of proteins that counteract oxidative stress, including enzymes that can scavenge and detoxify ROS induced by Cd. Moreover, B. licheniformis A can up-regulate energy transportation system components to meet the needs of their growth under heavy metal stress and achieve heavy metal tolerance. These indicate that B. licheniformis A is a promising microorganism for the experimental bioremediation of soils contaminated with Cd.Acknowledgements
This work was financially supported by State Oceanic Administration (201105007).References
- J. Shim, J.-M. Lim, P. J. Shea and B.-T. Oh, J. Hazard. Mater., 2014, 272, 129–136 CrossRef CAS PubMed.
- M. Filipič, Mutat. Res., 2012, 733, 69–77 CrossRef PubMed.
- A. Jamers, R. Blust, W. De Coen, J. L. Griffin and O. A. H. Jones, Aquat. Toxicol., 2013, 126, 355–364 CrossRef CAS PubMed.
- IARC, Beryllium, cadmium, mercury, and exposures in the glass manufacturing industry, World Health Organization, Lyon, France, 1993 Search PubMed.
- G. O. Oyetibo, M. O. Ilori, O. S. Obayori and O. O. Amund, Environ. Monit. Assess., 2013, 185, 6809–6818 CrossRef CAS PubMed.
- T. B. Ozer, I. A. Erkaya, A. U. Udoh, D. Y. Duygu, A. Akbulut, G. Bayramoglu and M. Y. Arica, Environ. Sci. Pollut. Res., 2012, 19, 2983–2993 CrossRef CAS PubMed.
- M. Cristani, C. Naccari, A. Nostro, A. Pizzimenti, D. Trombetta and F. Pizzimenti, Environ. Sci. Pollut. Res., 2012, 19, 161–168 CrossRef CAS PubMed.
- I. Poirier, P. Hammann, L. Kuhn and M. Bertrand, Aquat. Toxicol., 2013, 128-129, 215–232 CrossRef CAS PubMed.
- N. K. Kılıç, A. Stensballe, D. E. Otzen and G. Dönmez, Bioresour. Technol., 2010, 101, 2134–2140 CrossRef PubMed.
- R. Andreazza, S. Pieniz, L. Wolf, M.-K. Lee, F. A. O. Camargo and B. C. Okeke, Sci. Total Environ., 2010, 408, 1501–1507 CrossRef CAS PubMed.
- J. Y. Lee, J. G. Yang, D. Zhitnitsky, O. Lewinson and D. C. Rees, Science, 2014, 343, 1133–1136 CrossRef CAS PubMed.
- C. C. Wu, A. Gardarin, P. Catty, F. Guillain and E. Mintz, Biochimie, 2006, 88, 1687–1692 CrossRef CAS PubMed.
- Y. Song, S. Swift, P. J. Swedlund and N. Singhal, Appl. Geochem., 2011, 26, 898–906 CrossRef CAS PubMed.
- A. Manara, G. DalCorso, C. Baliardini, S. Farinati, D. Cecconi and A. Furini, J. Protein Res., 2012, 11, 4169–4179 CrossRef CAS PubMed.
- A. L. C. Mattos, V. R. L. Constantino, R. A. A. de Couto, D. M. L. Pinto, T. M. Kaneko and B. P. Espósito, J. Trace Elem. Med Biol., 2013, 27, 103–108 CAS.
- S. Pandey, P. Ghosh, S. Ghosh, T. De and T. Maiti, J. Microbiol., 2013, 51, 11–17 CrossRef CAS PubMed.
- I. V. N. Rathnayake, M. Megharaj, G. S. R. Krishnamurti, N. S. Bolan and R. Naidu, Chemosphere, 2013, 90, 1195–1200 CrossRef CAS PubMed.
- H. Wu, C. Ji, L. Wei, J. Zhao and H. Lu, J. Proteomics, 2013, 94, 54–67 CrossRef CAS PubMed.
- D. J. Spurgeon, O. A. H. Jones, J.-L. C. M. Dorne, C. Svendsen, S. Swain and S. R. Stürzenbaum, Sci. Total Environ., 2010, 408, 3725–3734 CrossRef CAS PubMed.
- T. Monsinjon and T. Knigge, Proteomics, 2007, 7, 2997–3009 CrossRef CAS PubMed.
- M. Aivaliotis, W. Haase, M. Karas and G. Tsiotis, Proteomics, 2006, 6, 217–232 CrossRef CAS PubMed.
- C. Bar, R. Patil, J. Doshi, M. J. Kulkarni and W. N. Gade, J. Biotechnol., 2007, 128, 444–451 CrossRef CAS PubMed.
- F. Zakeri, M. Sadeghizadeh, M. R. Kardan, H. Shahbani Zahiri, G. Ahmadian, F. Masoumi, H. Sharafi, G. Rigi, H. Vali and K. Akbari Noghabi, J. Proteomics, 2012, 75, 4820–4832 CrossRef CAS PubMed.
- S. Siripornadulsil, L. Thanwisai and W. Siripornadulsil, Can. J. Microbiol., 2014, 60, 121–131 CrossRef CAS PubMed.
- D. Mahle, P. Anderson, N. DelRaso, M. Raymer, A. Neuforth and N. Reo, Metabolomics, 2011, 7, 206–216 CrossRef CAS.
- G. Brunetti, K. Farrag, P. Soler-Rovira, M. Ferrara, F. Nigro and N. Senesi, Geoderma, 2012, 170, 322–330 CrossRef CAS PubMed.
- C. Dimkpa, T. Weinand and F. Asch, Plant, Cell Environ., 2009, 32, 1682–1694 CrossRef CAS PubMed.
- O. H. Jones, M. Maguire, J. Griffin, Y.-H. Jung, J. Shibato, R. Rakwal, G. Agrawal and N.-S. Jwa, Eur. J. Plant Pathol., 2011, 129, 539–554 CrossRef CAS.
- J. E. Le Belle, N. G. Harris, S. R. Williams and K. K. Bhakoo, NMR Biomed., 2002, 15, 37–44 CrossRef CAS PubMed.
- C. M. Slupsky and B. D. Sykes, Metabolomics, 2006, 2, 113–123 CrossRef.
- M. Neerathilingam, D. E. Volk, S. Sarkar, T. M. Alam, M. K. Alam, G. A. S. Ansari and B. A. Luxon, Toxicol. Lett., 2010, 199, 10–16 CrossRef CAS PubMed.
- N. D. Meadow, D. K. Fox and S. Roseman, Annu. Rev. Biochem., 1990, 59, 497–542 CrossRef CAS PubMed.
- M. J. Danson, R. Eisenthal, S. Hall, S. R. Kessell and D. L. Williams, Biochem. J., 1984, 218, 811–818 CAS.
- A. Rince, S. Flahaut and Y. Auffray, Int. J. Food Microbiol., 2000, 55, 87–91 CrossRef CAS.
- M. Gamerdinger and E. Deuerling, Science, 2014, 344, 590–591 CrossRef CAS PubMed.
- S. Tang, Q. Cai, H. Chibli, V. Allagadda, J. L. Nadeau and G. D. Mayer, Toxicol. Appl. Pharmacol., 2013, 272, 443–452 CrossRef CAS PubMed.
- D. N. Wilson, Nat. Rev. Microbiol., 2014, 12, 35–48 CrossRef CAS PubMed.
- S. Sharma, C. S. Sundaram, P. M. Luthra, Y. Singh, R. Sirdeshmukh and W. N. Gade, J. Biotechnol., 2006, 126, 374–382 CrossRef CAS PubMed.
- R. Schroeter, B. Voigt, B. Jürgen, K. Methling, D.-C. Pöther, H. Schäfer, D. Albrecht, J. Mostertz, U. Mäder, S. Evers, K.-H. Maurer, M. Lalk, T. Mascher, M. Hecker and T. Schweder, Proteomics, 2011, 11, 2851–2866 CrossRef CAS PubMed.
- M. J. Penninckx and M. T. Elskens, Adv. Microb. Physiol., 1993, 34, 239–301 CrossRef CAS.
- L.-L. Lin, Y.-Y. Chen, M.-C. Chi and A. Merlino, Biochim. Biophys. Acta, Proteins Proteomics, 2014, 1844, 1523–1529 CrossRef CAS PubMed.
- E. Bianucci, A. Fabra and S. Castro, Curr. Microbiol., 2011, 62, 96–100 CrossRef CAS PubMed.
- A. Bignucolo, V. P. Appanna, S. C. Thomas, C. Auger, S. Han, A. Omri and V. D. Appanna, J. Biotechnol., 2013, 167, 309–315 CrossRef CAS PubMed.
- C. Sohlenkamp, I. M. López-Lara and O. Geiger, Prog. Lipid Res., 2003, 42, 115–162 CrossRef CAS.
Footnote |
| † Present address: Fenghua Road, Ningbo City, Zhejiang Province 315211, P. R. China. |
| This journal is © The Royal Society of Chemistry 2015 |