
Novel photovoltaic donor 1–acceptor–donor 2–acceptor terpolymers with tunable energy levels based on a difluorinated benzothiadiazole acceptor†
Abstract
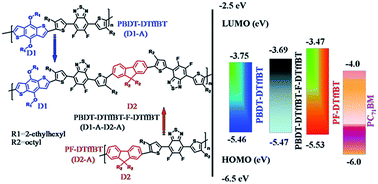
A novel donor 1–acceptor–donor 2–acceptor (D1–A–D2–A) type terpolymer PBDT-DTffBT-F-DTffBT was prepared to further tune the energy levels of donor–acceptor (D–A) type copolymer PBDT-DTffBT and PF-DTffBT, and the corresponding optoelectronic properties have been investigated. By incorporating the weak donor fluorene unit into the backbone of PBDT-DTffBT, the PBDT-DTffBT-F-DTffBT exhibits a lower HOMO level and higher LUMO level compared with PBDT-DTffBT as expected. The polymer solar cell (PSC) based on PBDT-DTffBT-F-DTffBT affords the improved PCE with a higher Voc of 0.853 V compared to the D–A copolymer PBDT-DTffBT and PF-DTffBT. Therefore, through carefully choosing the suitable donor group, the class of D1–A–D2–A type copolymers would be a promising organic semiconducting material.
 Please wait while we load your content...
Please wait while we load your content...

