
Bionanotherapeutics: niclosamide encapsulated albumin nanoparticles as a novel drug delivery system for cancer therapy†
Abstract
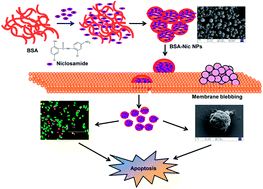
One of the major unresolved challenges among the scientific community is to develop anticancer drugs that are safe and effective. A large number of anticancer drugs have been screened so far in this campaign. Among them niclosamide has shown tremendous anti-cancer potential as demonstrated in a surfeit of human cancer cell lines and animal models. But the extreme hydrophobicity and consequently, minimal systemic bioavailability associated with this drug limited its widespread clinical applications. Nanoparticles based drug delivery systems have the potential for realizing water soluble formulation of highly hydrophobic anticancer drugs like niclosamide, thus evading the drawbacks of poor solubility. In this work niclosamide was encapsulated into albumin nanoparticles through a desolvation method to improve its scope of application in cancer therapy. Physico-chemical characterization confirms that the prepared nanoparticles are spherical, highly monodispersed, and stable in aqueous systems. These drug encapsulated albumin nanoparticles, unlike the free drug demonstrate better in vitro therapeutic efficacy against human lung and breast cancer cell lines, as assessed by cell viability assay and morphological analyses. Further, the proficient induction of apoptosis by these nanoparticles was confirmed by semi-quantitative RT-PCR. This work open up a new avenue to extend the clinical gamut of this effectual agent by enabling its aqueous dispersion.
 Please wait while we load your content...
Please wait while we load your content...

