
Oxidation of SO2 and NO by epoxy groups on graphene oxides: the role of the hydroxyl group†
Abstract
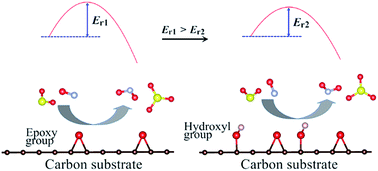
Simultaneous catalytic removal of SO2 and NOx at low temperature (<150 °C) has long been recognized as a challenge for the treatment of coal-burned flue gases. Density functional theory corrected with dispersion was used to investigate the potential of graphene oxides (GOs) for the catalytic oxidation of SO2 and NOx. It is found that both the SO2 and NOx can be oxidized by epoxy groups of GO nearly at room temperature. The hydroxyl groups on the GO surface enhance the adsorption and oxidation of SO2, and of NO as well, but in quite different ways. For the case of SO2, the promotion is derived from the formation of charge transfer channels, which are fabricated by the hydroxyl group, the adsorbed SO2 and the epoxy group. The promotion is enhanced by the introduction of more hydroxyl groups as more charge transfer channels are formed. However, for NO, the hydroxyl group leads to a strong N–C covalent interaction between the adsorbed NO molecules and the GO surface, through which the NO is activated and oxidized with a much lower barrier. These results provide a mechanistic explanation of the low temperature catalytic oxidation of SO2 and NO by carbon materials and insights into designing new carbon-based catalysts.
 Please wait while we load your content...
Please wait while we load your content...

